
Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction


Abstract
:
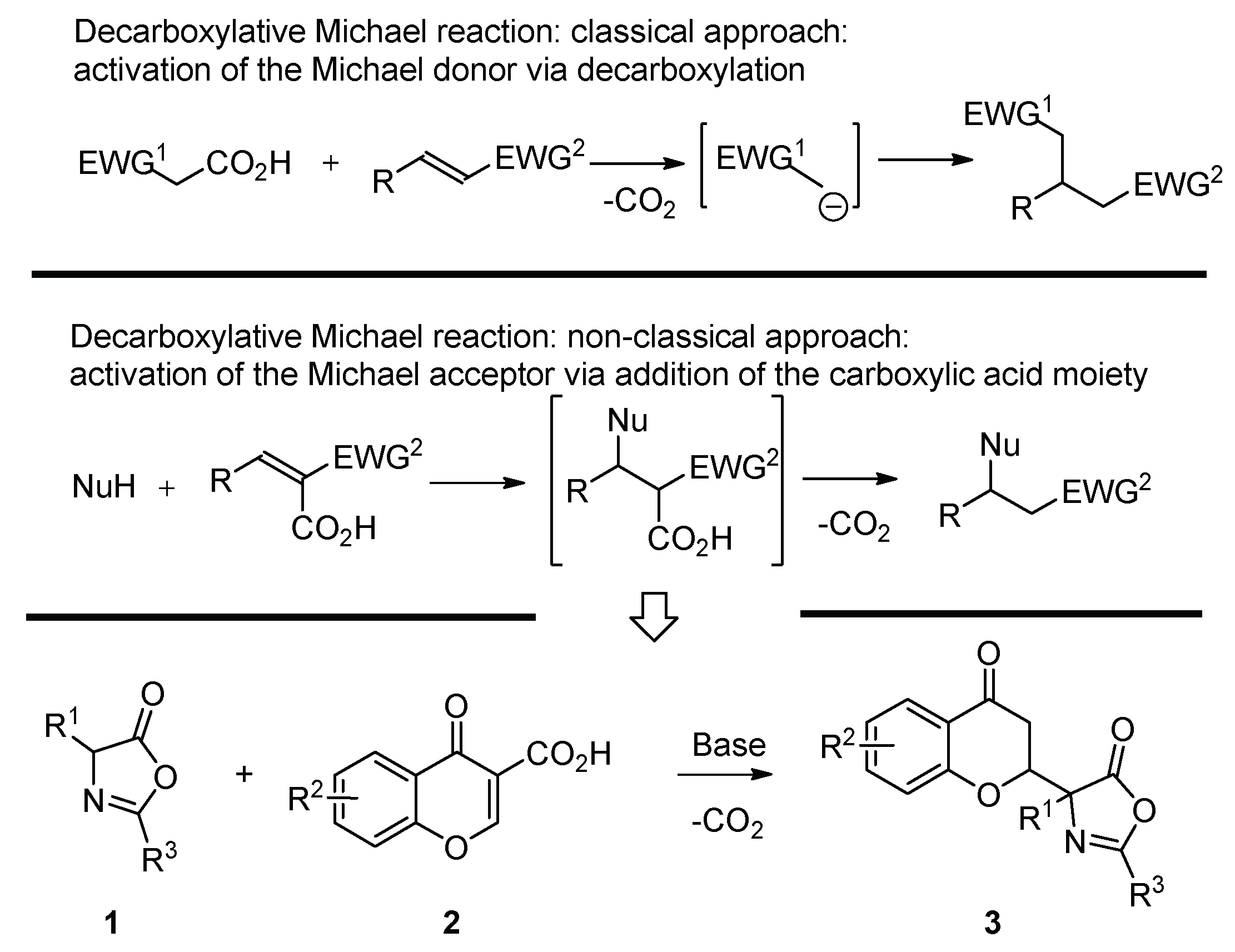
1. Introduction
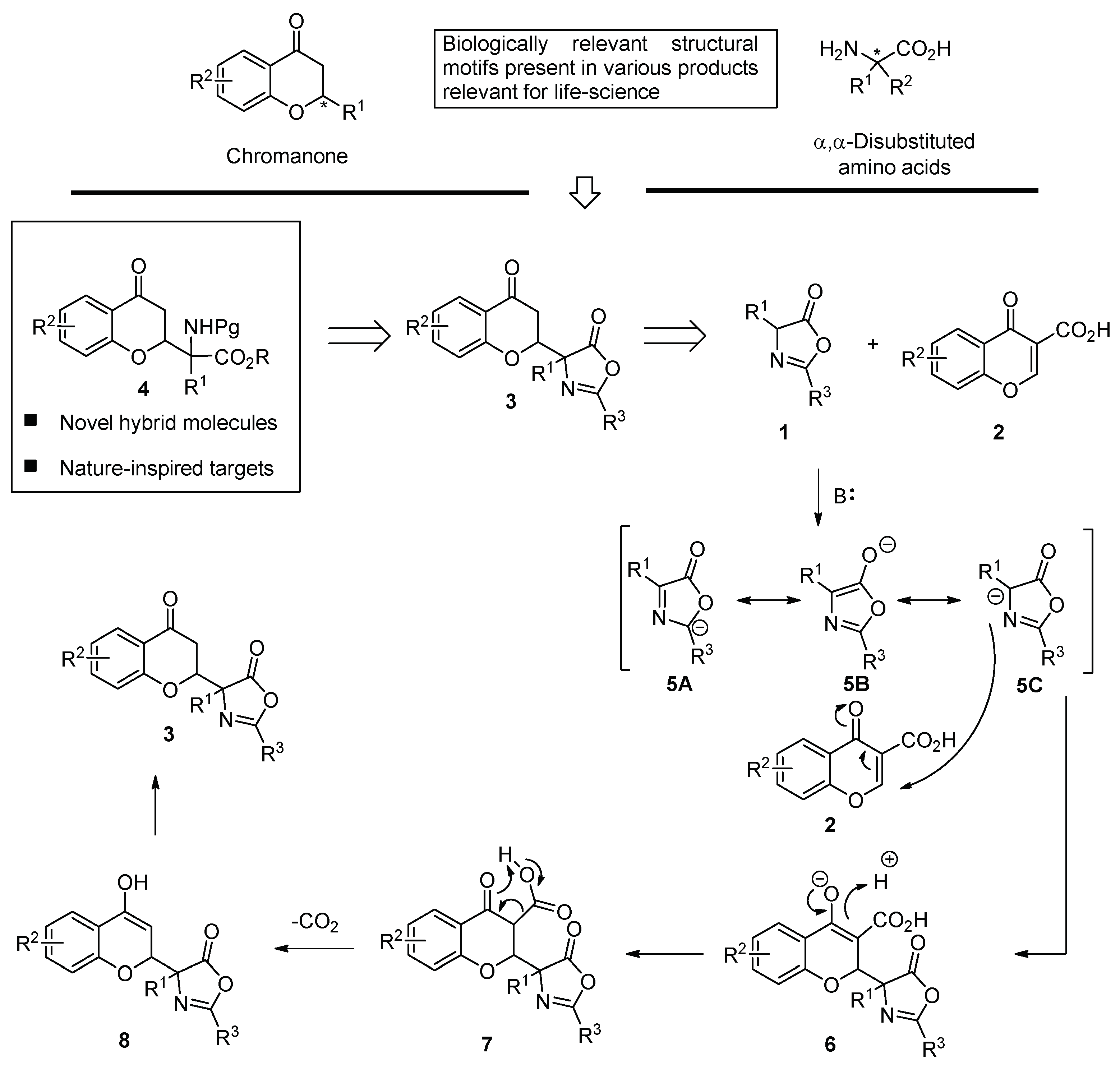
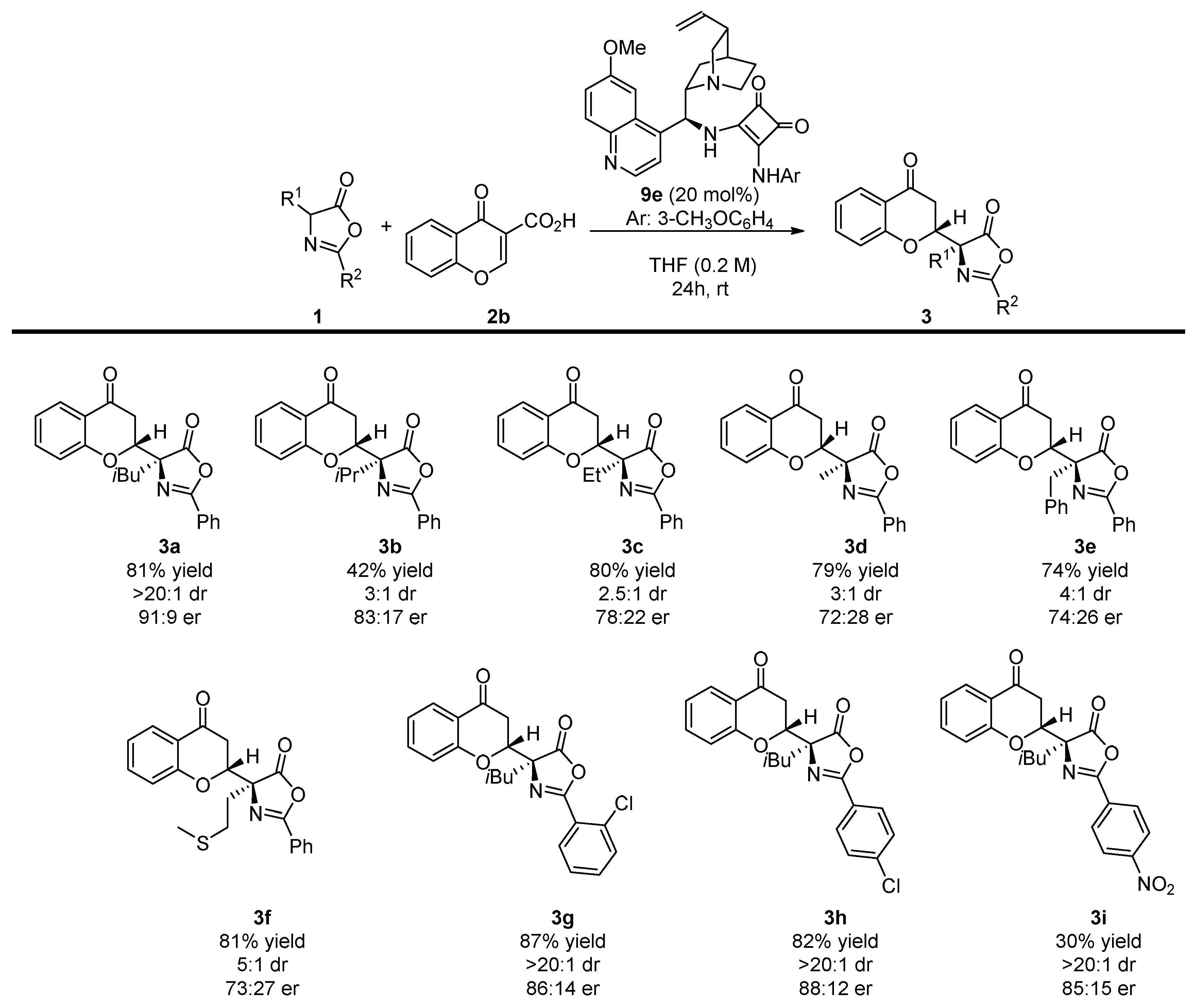
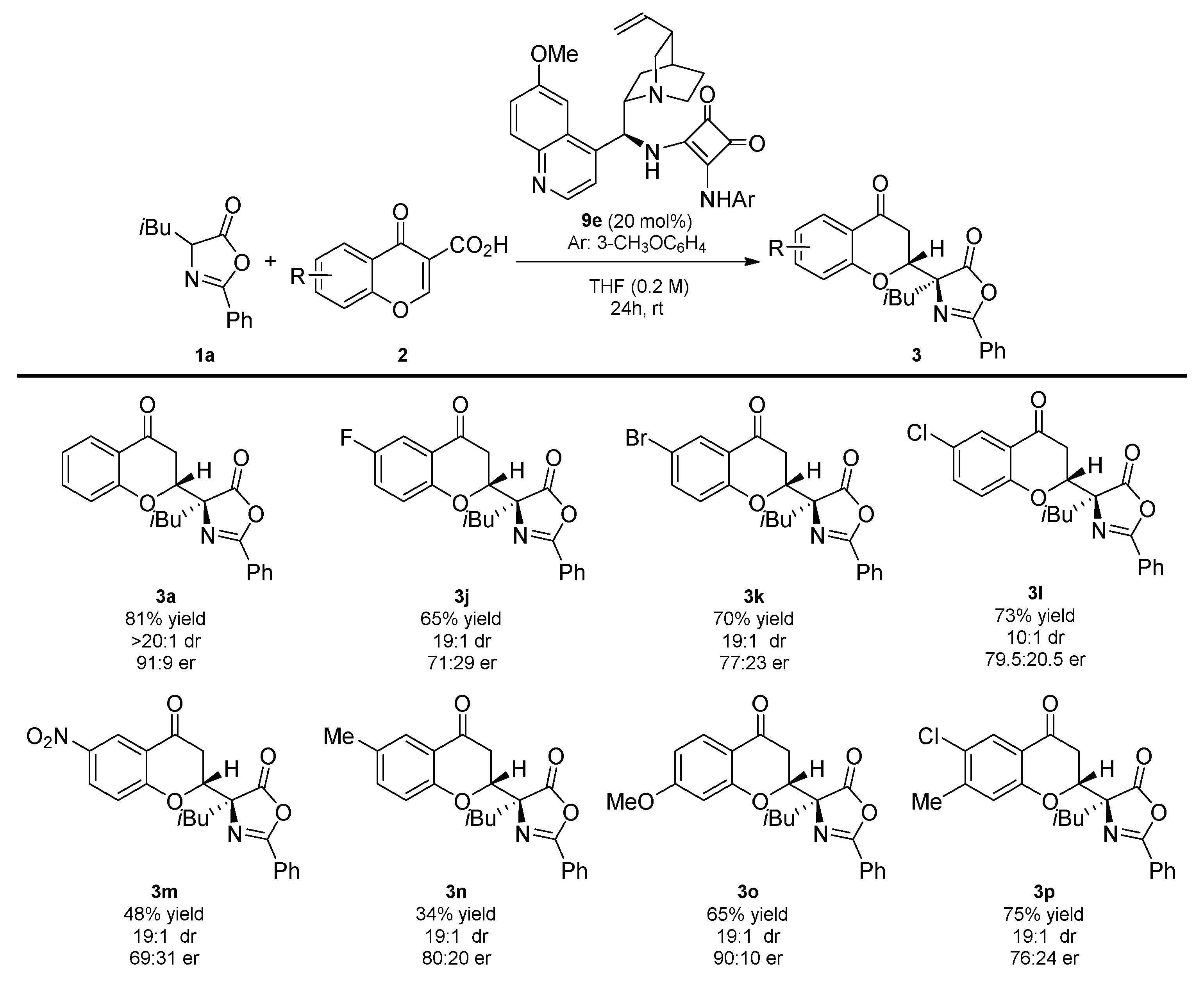
2. Results
3. Conclusion
4. Materials and Methods
4.1. General Methods
4.2. General Procedure
4.3. Synthesis of Methyl 2-Benzamido-4-Methyl-2-(4-Oxochroman-2-yl)Pentanoate (4a)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Sibi, M.P.; Manyem, S. Enantioselective Conjugate Additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Röder, A. Recent Advances in Catalytic Enantioselective Michael Additions. Synthesis 2001, 2, 171–196. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael Additions to Nitroalkenes. Eur. J. Org. Chem. 2002, 12, 1877–1894. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Construction of Quaternary Stereocenters: New Perspectives through Enantioselective Michael Reactions. Angew. Chem. Int. Ed. 2003, 42, 1688–1690. [Google Scholar] [CrossRef] [PubMed]
- Christoffers, J.; Koripelly, G.; Rosiak, A.; Rössle, M. Recent Advances in Metal-Catalyzed Asymmetric Conjugate Additions. Synthesis 2007, 9, 1279–1300. [Google Scholar] [CrossRef]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic Enantioselective Michael and Hetero-Michael Reactions. Synthesis 2007, 14, 2065–2092. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 11, 1701–1716. [Google Scholar] [CrossRef]
- Almasi, D.; Alonso, D.A.; Nájera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Schwarz, J.; König, B. Decarboxylative reactions with and without light—A comparison. Green Chem. 2018, 20, 323–361. [Google Scholar] [CrossRef]
- Pan, Y.; Tan, C.H. Catalytic Decarboxylative Reactions: Biomimetic Approaches Inspired by Polyketide Biosynthesis. Synthesis 2011, 13, 2044–2053. [Google Scholar] [CrossRef]
- Wang, Z.L. Recent Advances in Catalytic Asymmetric Decarboxylative Addition Reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Bojanowski, J.; Albrecht, A. Carboxylic-acid-activated olefins in decarboxylative reactions. Asian J. Org. Chem. 2019, 8, 746–754. [Google Scholar] [CrossRef]
- Lubkoll, J.; Wennemers, H. Mimicry of Polyketide Synthases—Enantioselective 1,4-Addition Reactions of Malonic Acid Half-Thioesters to Nitroolefins. Angew. Chem. Int. Ed. 2007, 46, 6841–6844. [Google Scholar] [CrossRef]
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A Heterobimetallic Ni/La-salan Complex for Catalytic Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half-Thioester. Chem. Asian J. 2010, 15, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to β-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of γ-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202. [Google Scholar] [CrossRef]
- Kang, Y.K.; Lee, H.J.; Moon, H.W.; Kim, D.Y. Organocatalytic enantioselective decarboxylative Michael addition of β-ketoacids to α,β-unsaturated ketones. RSC Adv. 2013, 3, 1332–1335. [Google Scholar] [CrossRef]
- Qiao, B.; Liu, Q.; Liu, H.; Yan, L.; Jiang, Z. Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half Thioesters to Vinyl Sulfones: Highly Enantioselective Synthesis of 3-Monofluoromethyl-3-Arylpropanoic Esters. Chem. Asian J. 2014, 9, 1252–1256. [Google Scholar] [CrossRef]
- Ren, Q.; Sun, S.; Huang, J.; Li, W.; Wu, M.; Guo, H.; Wang, J. An enantioselective cascade reaction between α,β-unsaturated aldehydes and malonic half-thioesters: A rapid access to chiral δ-lactones. Chem. Commun. 2014, 50, 6137–6140. [Google Scholar] [CrossRef]
- Ren, Q.; Gao, T.; Li, W.; Wan, L.; Hu, Y.; Peng, Y.; Sun, S.; Hu, L.; Wu, M.; Guo, H.; et al. A highly enantioselective Michael reaction between α,β-unsaturated ketones and malonic acid half-thioesters. New. J. Chem. 2015, 39, 5100–5103. [Google Scholar] [CrossRef]
- Nakamura, S.; Toda, A.; Sano, M.; Hatanaka, T.; Funahashi, Y. Organocatalytic Enantioselective Conjugate Addition of Malonic Acid Half Thioesters to Coumarin-3-carboxylic Acids Using N-Heteroarenesulfonyl Cinchona Alkaloid Amides. Adv. Synth. Catal. 2016, 358, 1029–1034. [Google Scholar] [CrossRef]
- Brunner, H.; Baur, M.A. α-Amino Acid Derivatives by Enantioselective Decarboxylation. Eur. J. Org. Chem. 2003, 15, 2854–2862. [Google Scholar] [CrossRef]
- Rogers, L.M.A.; Rouden, J.; Lecomte, L.; Lasne, M.C. Enantioselective decarboxylation–reprotonation of an α-amino malonate derivative as a route to optically enriched cyclic α-amino acid. Tetrahedron Lett. 2003, 44, 3047–3050. [Google Scholar] [CrossRef]
- Seitz, T.; Baudoux, J.; Bekolo, H.; Cahard, D.; Plaquevent, J.C.; Lasne, M.C.; Rouden, J. Organocatalyzed route to enantioenriched pipecolic esters: Decarboxylation of an aminomalonate hemiester. Tetrahedron 2006, 62, 6155–6165. [Google Scholar] [CrossRef]
- Amere, M.; Lasne, M.C.; Rouden, J. Highly Enantioselective Decarboxylative Protonation of α-Aminomalonates Mediated by Thiourea Cinchona Alkaloid Derivatives: Access to Both Enantiomers of Cyclic and Acyclic α-Aminoacids. Org. Lett. 2007, 9, 2621–2624. [Google Scholar] [CrossRef]
- Yuan, H.N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J.A. Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083. Angew. Chem. Int. Ed. 2013, 52, 3869–3873. [Google Scholar] [CrossRef]
- Wei, A.J.; Nie, J.; Zheng, Y.; Ma, J.A. Ni-Catalyzed Highly Chemo-, Regio-, and Enantioselective Decarboxylative Aldol Reaction of β,γ-Unsaturated α-Ketoesters with β-Ketoacids. J. Org. Chem. 2015, 80, 3766–3776. [Google Scholar] [CrossRef]
- Wei, Y.; Guo, R.; Dang, Y.; Nie, J.; Ma, J.A. Organocatalytic Enantioselective Decarboxylative Michael Addition of β-Keto Acids to Dicyanoolefins and Disulfonylolefins. Adv. Synth. Catal. 2016, 358, 2721–2726. [Google Scholar] [CrossRef]
- Wallace, T.W. Conjugate addition to chromones: Synthesis of substituted 4-chromanones. Tetrahedron Lett. 1984, 25, 4299–4302. [Google Scholar] [CrossRef]
- Neo, A.G.; Díaz, J.; Marcaccini, S.; Marcos, C.F. Conjugate addition of isocyanides to chromone 3-carboxylic acid: An efficient one-pot synthesis of chroman-4-one 2-carboxamides. Org. Biomol. Chem. 2012, 10, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, L.; Xu, L.; Zhao, H.; Xiao, J. Facile synthesis of azaarene-2-substituted chromanone derivatives via tandem sp3 C–H functionalization/decarboxylation of azaarenes with 4-oxo-4H-chromene-3-carboxylic acid. RSC Adv. 2014, 4, 53188–53191. [Google Scholar] [CrossRef]
- Xu, L.; Shao, Z.; Wang, L.; Xiao, J. Tandem sp3 C–H Functionlization/Decarboxylation of 2-Alkylazaarenes with Coumarin-3-carboxylic Acids. Org. Lett. 2014, 16, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Xu, L.; Wang, L.; Wie, H.; Xiao, J. Catalyst-free tandem Michael addition/decarboxylation of (thio)coumarin-3-carboxylic acids with indoles: Facile synthesis of indole-3-substituted 3,4-dihydro(thio)coumarins. Org. Biomol. Chem. 2014, 12, 2185–2188. [Google Scholar] [CrossRef] [PubMed]
- Talhi, O.; Brodziak-Jarosz, L.; Panning, J.; Orlikova, B.; Zwergel, C.; Tzanova, T.; Philippot, S.; Pinto, D.C.G.A.; Almeida Paz, F.A.; Gerhäuser, C.; et al. One-Pot Synthesis of Benzopyran-4-ones with Cancer Preventive and Therapeutic Potential. Eur. J. Org. Chem. 2016, 965–975. [Google Scholar] [CrossRef]
- Peng, S.; Wang, L.; Guo, H.; Sun, S.; Wang, J. Facile synthesis of 4-substituted 3,4-dihydrocoumarins via an organocatalytic double decarboxylation process. Org. Biomol. Chem. 2012, 10, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A. Utilization of Chromone-3-Carboxylic Acids as Acceptors in the Michael-Type Decarboxylative Addition. Eur. J. Org. Chem. 2018, 46, 6482–6485. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Applications; Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Saengchantara, S.T.; Wallace, T.W. Chromanols, chromanones, and chromones. Nat. Prod. Rep. 1986, 3, 465–475. [Google Scholar] [CrossRef]
- Kamat, D.P.; Tilve, S.G.; Kamat, V.P.; Kirtany, J.K. Syntheses and Biological Activities of Chroman-2-ones. A Review. Org. Prep. Proc. Int. 2015, 47, 1–79. [Google Scholar] [CrossRef]
- Masters, K.S.; Bräse, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Eggleston, D.S.; Haltiwanger, R.C.; Bean, M.F.; Taylor, P.B.; Caranfa, M.J.; Breen, A.L.; Bartus, H.R.; Johnson, R.K. The inophyllums, novel inhibitors of HIV-1 reverse transcriptase isolated from the Malaysian tree, Calophyllum inophyllum Linn. J. Med. Chem. 1993, 36, 4131–4138. [Google Scholar] [CrossRef] [PubMed]
- Galinis, D.L.; Fuller, R.W.; McKee, T.C.; Cardellina, J.H., II; Gulakowski, R.J.; McMahon, J.B.; Boyd, M.R. Structure−Activity Modifications of the HIV-1 Inhibitors (+)-Calanolide A and (−)-Calanolide B. J. Med. Chem. 1996, 39, 4507–4510. [Google Scholar] [CrossRef] [PubMed]
- Mbwambo, Z.H.; Kapingu, M.C.; Moshi, M.J.; Machumi, F.; Apers, S.; Cos, P.; Ferreira, D.; Marais, J.P.J.; Berghe, D.V.; Maes, L.; et al. Antiparasitic Activity of Some Xanthones and Biflavonoids from the Root Bark of Garcinia livingstonei. J. Nat. Prod. 2006, 69, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Picker, K.; Ritchie, E.; Taylor, W.C. The chemical constituents of Australian Flindersia species. XXI. An examination of the bark and the leaves of F. laevicarpa. Aust. J. Chem. 1976, 29, 2023–2036. [Google Scholar] [CrossRef]
- Zhao, D.L.; Shao, C.L.; Gan, L.S.; Wang, M.; Wang, C.Y. Chromone Derivatives from a Sponge-Derived Strain of the Fungus Corynespora cassiicola. J. Nat. Prod. 2015, 78, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, A.E.; Scheidt, K.A. Asymmetric Methods for the Synthesis of Flavanones, Chromanones, and Azaflavanones. Eur. J. Org. Chem. 2012, 449–462. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.R.; Scheidt, K.A. Pyranone Natural Products as Inspirations for Catalytic Reaction Discovery and Development. Acc. Chem. Res. 2015, 48, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, A.; Bojanowski, J. Decarboxylative Aminocatalytic Cascade for the Synthesis of Dihydroxanthones. Adv. Synth. Catal. 2017, 358, 2907–2911. [Google Scholar] [CrossRef]
- Wen, G.; Su, Y.; Zhang, G.; Lin, Q.; Zhu, Y.; Zhang, Q.; Fang, X. Stereodivergent Synthesis of Chromanones and Flavanones via Intramolecular Benzoin Reaction. Org. Lett. 2016, 18, 3980–3983. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. (−)-β-Pinene-Derived N-Heterocyclic Carbenes: Application to Highly Enantioselective Intramolecular Stetter Reaction. ACS Catal. 2014, 4, 1404–1408. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A. Enantioselective Synthesis of Chromanones Bearing Quaternary Substituted Stereocenters Catalyzed by (1R)-Camphor-Derived N-Heterocyclic Carbenes. J. Org. Chem. 2015, 80, 7468–7476. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.Q.; Li, Y.; Yang, G.Q.; You, S.L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett 2011, 1033–1037. [Google Scholar] [CrossRef]
- Biddle, M.M.; Lin, M.; Scheidt, K.A. Catalytic Enantioselective Synthesis of Flavanones and Chromanones. J. Am. Chem. Soc. 2007, 129, 3830–3831. [Google Scholar] [CrossRef] [PubMed]
- Read de Alaniz, J.; Rovis, T.A. Highly Enantio- and Diastereoselective Catalytic Intramolecular Stetter Reaction. J. Am. Chem. Soc. 2005, 127, 6284–6289. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Runsink, J. The First Asymmetric Intramolecular Stetter Reaction. Preliminary Communication. Helv. Chim. Acta 1996, 79, 1899–1902. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D. Stereoselective synthesis of quaternary α-amino acids. Part 2: Cyclic compounds. Tetrahedron Asymmetry 2000, 11, 645–732. [Google Scholar] [CrossRef]
- Vogt, H.; Bräse, S. Recent approaches towards the asymmetric synthesis of α,α-disubstituted α-amino acids. Org. Biomol. Chem. 2007, 5, 406–430. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D. Recent progress on the stereoselective synthesis of acyclic quaternary α-amino acids. Tetrahedron Asymmetry 2007, 18, 569–623. [Google Scholar] [CrossRef]
- Ohfune, Y.; Shinada, T. Enantio and Diastereoselective Construction of α,α-Disubstituted α-Amino Acids for the Synthesis of Biologically Active Compounds. Eur. J. Org. Chem. 2005, 5127–5143. [Google Scholar] [CrossRef]
- Nájera, C. From α-Amino Acids to Peptides: All You Need for the Journey. Synlett 2002, 9, 1388–1404. [Google Scholar] [CrossRef]
- Alba, A.N.; Rios, R. Oxazolones in Organocatalysis, New Tricks for an Old Reagent. Chem. Asian. J. 2011, 6, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Scala, A.; Risitano, F.; Grassi, G. Oxazol-5-(4H)-Ones. Part 1. Synthesis and Reactivity as 1,3-dipoles. Curr. Org. Chem. 2014, 18, 2691–2710. [Google Scholar] [CrossRef]
- De Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Reyes, E.; Alemán, J.; Milelli, A.; Kobbelgaard, S.; Jørgensen, K.A. Organocatalytic Asymmetric Synthesis of α,α-Disubstituted α-Amino Acids and Derivatives. J. Am. Chem. Soc. 2008, 130, 12031–12037. [Google Scholar] [CrossRef] [PubMed]
- Balaguer, A.N.; Companyó, X.; Calvet, T.; Font-Bardia, M.; Moyano, A.; Rios, R. Highly Regio and Diastereoselective Oxazol-5-one Addition to Nitrostyrenes. Eur. J. Org. Chem. 2009, 199–203. [Google Scholar] [CrossRef]
- Alba, A.N.R.; Companyó, X.; Valero, G.; Moyano, A.; Rios, R. Enantioselective Organocatalytic Addition of Oxazolones to 1,1-Bis(phenylsulfonyl)ethylene: A Convenient Asymmetric Synthesis of Quaternary α-Amino Acids. Chem. Eur. J. 2010, 16, 5354–5361. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Dzięgielewski, M.; Kowalczyk, D.; Albrecht, L. A Convenient Approach to a Novel Group of Quaternary Amino Acids Containing a Geminal Bisphosphonate Moiety. Synthesis 2014, 46, 3233–3238. [Google Scholar] [CrossRef]
- Jiang, H.; Gschwend, B.; Albrecht, L.; Hansen, S.G.; Jørgensen, K.A. Asymmetric Trienamine Catalysis for the Construction of Structurally Rigid Cyclic α,α-Disubstituted Amino Acid Derivatives. Chem. Eur. J. 2011, 17, 9032–9036. [Google Scholar] [CrossRef]
- Marcelli, T.; van Maarseveen, J.H.; Hiemstra, H. Cupreines and Cupreidines: An Emerging Class of Bifunctional Cinchona Organocatalysts. Angew. Chem. Int. Ed. 2006, 45, 7496–7504. [Google Scholar] [CrossRef] [PubMed]
- Jew, S.S.; Park, H.G. Cinchona-based phase-transfer catalysts for asymmetric synthesis. Chem. Commun. 2009, 46, 7090–7103. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Oiarbide, M.; López, R. Asymmetric organocatalysis by chiral Brønsted bases: Implications and applications. Chem. Soc. Rev. 2009, 38, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.; Rodríguez-Docampo, Z.; Connon, S.J. Highly tunable arylated cinchona alkaloids as bifunctional catalysts. Chem. Commun. 2012, 48, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, H.; Dzięgielewski, M.; Deredas, D.; Albrecht, A.; Albrecht, L. Chiral Iminophosphoranes—An Emerging Class of Superbase Organocatalysts. Chem. Eur. J. 2015, 21, 10268–10277. [Google Scholar] [CrossRef] [PubMed]
- For the synthesis of catalyst 9e see: Bera, K.; Namboothiri, I.N.N. Quinine-Derived Thiourea and Squaramide Catalyzed Conjugate Addition of α-Nitrophosphonates to Enones: Asymmetric Synthesis of Quaternary α-Aminophosphonates. J. Org. Chem. 2015, 80, 1402–1413. [Google Scholar] [CrossRef] Analytical data of 9e was in accordance with the literature. Purity was confirmed by HRMS analysis: Calculated for [C31H33N4O4+H+]: 525.2496, found: 525.2507.
- CCDC 1895323 Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge from The Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/structure (accessed on 1 June 2019).
- Liang, J.; Ruble, J.C.; Fu, G.C. Dynamic Kinetic Resolutions Catalyzed by a Planar-Chiral Derivative of DMAP: Enantioselective Synthesis of Protected α-Amino Acids from Racemic Azlactones. J. Org. Chem. 1998, 63, 3154–3155. [Google Scholar] [CrossRef]
- Ishizuka, N.; Matsunori, K.; Sakai, K.; Fujimoto, M.; Mihara, S.; Yamamori, T. Structure−Activity Relationships of a Novel Class of Endothelin-A Receptor Antagonists and Discovery of Potent and Selective Receptor Antagonist, 2-(Benzo[1,3]dioxol-5-yl)-6-isopropyloxy-4-(4-methoxyphenyl)-2H-chromene-3- carboxylic Acid (S-1255). 1. Study on Structure−Activity Relationships and Basic Structure Crucial for ETA Antagonism. J. Med. Chem. 2002, 45, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
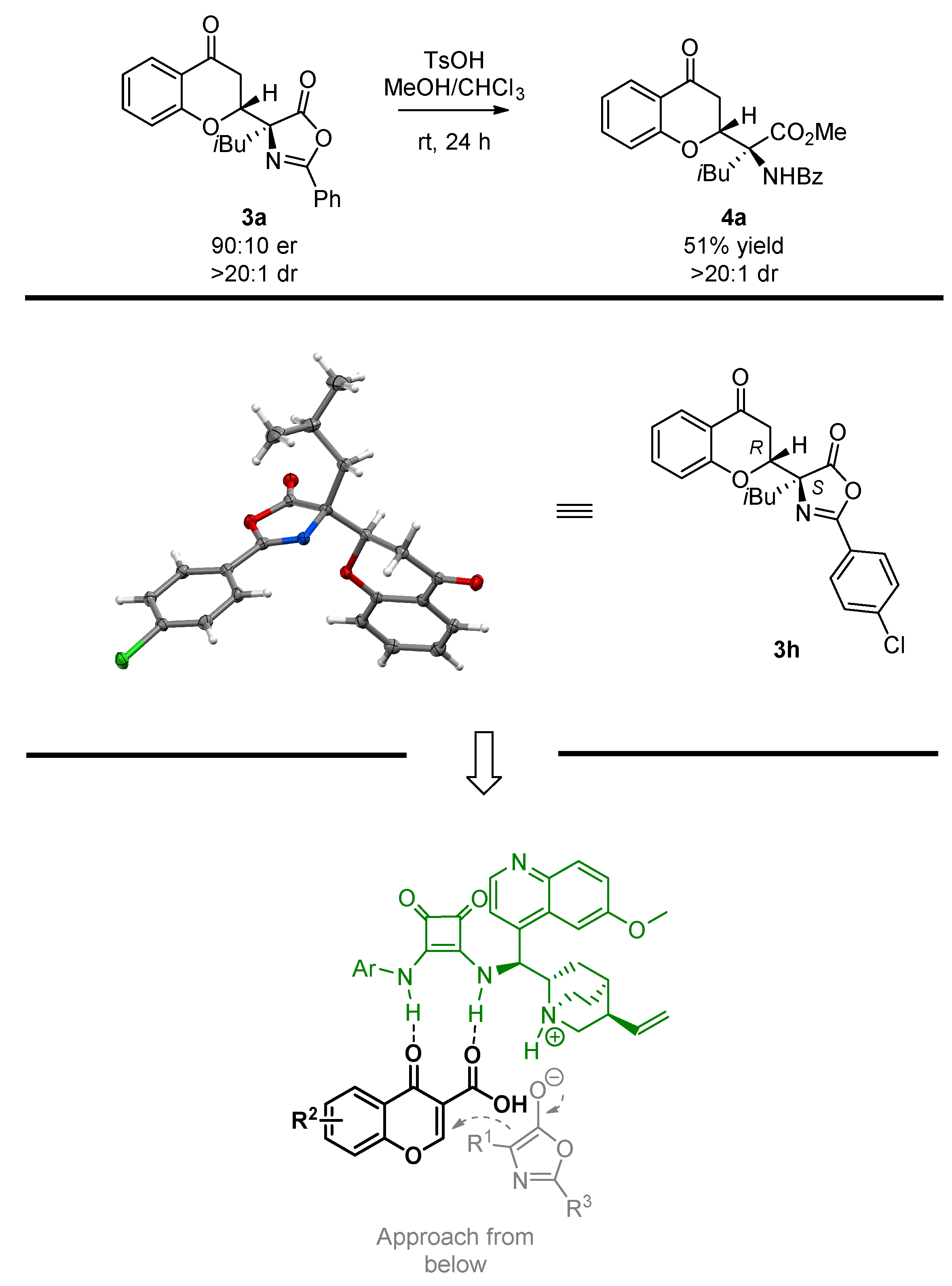
Sample Availability: Samples of the compounds 3a, 3h are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Solvent | 2/9 | Conv. (Yield) [%] b | dr c | er d | ee d [%] | |
|---|---|---|---|---|---|---|
| 1 | THF | 2a/9a | <5 | n.d. | n.d. | n.d. |
| 2 | THF | 2b/9a | >95 | 3:1 | 55:45 | 10 |
| 3 | THF | 2b/9b | >95 | 5:1 | 75:25 | 50 |
| 4 | THF | 2b/9c | >95 | 3:1 | 84:16 | 68 |
| 5 | THF | 2b/9d | >95 | 10:1 | 84:16 | 68 |
| 6 | THF | 2b/9e | >95 (81) | >20:1 | 91:9 | 82 |
| 7 | THF | 2b/9f | >95 | >20:1 | 79:21 | 58 |
| 8 | CH2Cl2 | 2b/9e | >95 | 5:1 | 81:19 | 62 |
| 9 | Toluene | 2b/9e | >95 | 5:1 | 91:9 | 82 |
| 10 | 1,4-Dioxane | 2b/9e | >95 | 10:1 | 87:13 | 74 |
| 11 | CPME | 2b/9e | >95 | 4:1 | 84:16 | 68 |
| 12 | Et2O | 2b/9e | >95 | 6:1 | 70:30 | 40 |
| 13 | 2-MeTHF | 2b/9e | >95 | 15:1 | 89:11 | 78 |
| 14 e | THF | 2b/9e | >95 | 20:1 | 90:10 | 80 |
| 15 f | THF | 2b/9e | >95 | 20:1 | 90:10 | 80 |
| 16 g | THF | 2b/9e | >95 | 10:1 | 70:30 | 40 |
| 17 h | THF | 2b/9e | >95 | 20:1 | 84:16 | 68 |
| 18 i | THF | 2b/9e | >95 | 20:1 | 84:16 | 68 |
| 19 j | THF | 2b/9e | >95 (76) | >20:1 | 91:9 | 82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojanowski, J.; Sieroń, L.; Albrecht, A. Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules 2019, 24, 2565. https://doi.org/10.3390/molecules24142565
Bojanowski J, Sieroń L, Albrecht A. Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules. 2019; 24(14):2565. https://doi.org/10.3390/molecules24142565
Chicago/Turabian StyleBojanowski, Jan, Lesław Sieroń, and Anna Albrecht. 2019. "Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction" Molecules 24, no. 14: 2565. https://doi.org/10.3390/molecules24142565
APA StyleBojanowski, J., Sieroń, L., & Albrecht, A. (2019). Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules, 24(14), 2565. https://doi.org/10.3390/molecules24142565