
Comparison of Various Chromatographic Systems for Analysis of Cytisine in Human Serum, Saliva and Pharmaceutical Formulation by HPLC with Diode Array, Fluorescence or Mass Spectrometry Detection

 ,
,  ,
,  ,
,
Abstract
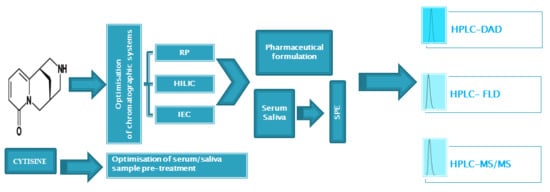
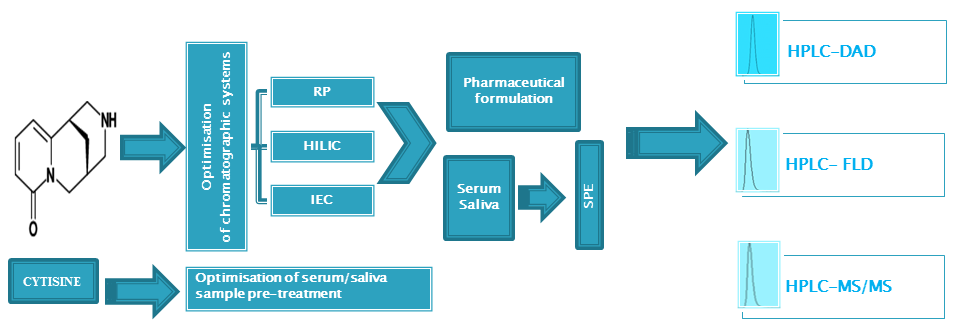
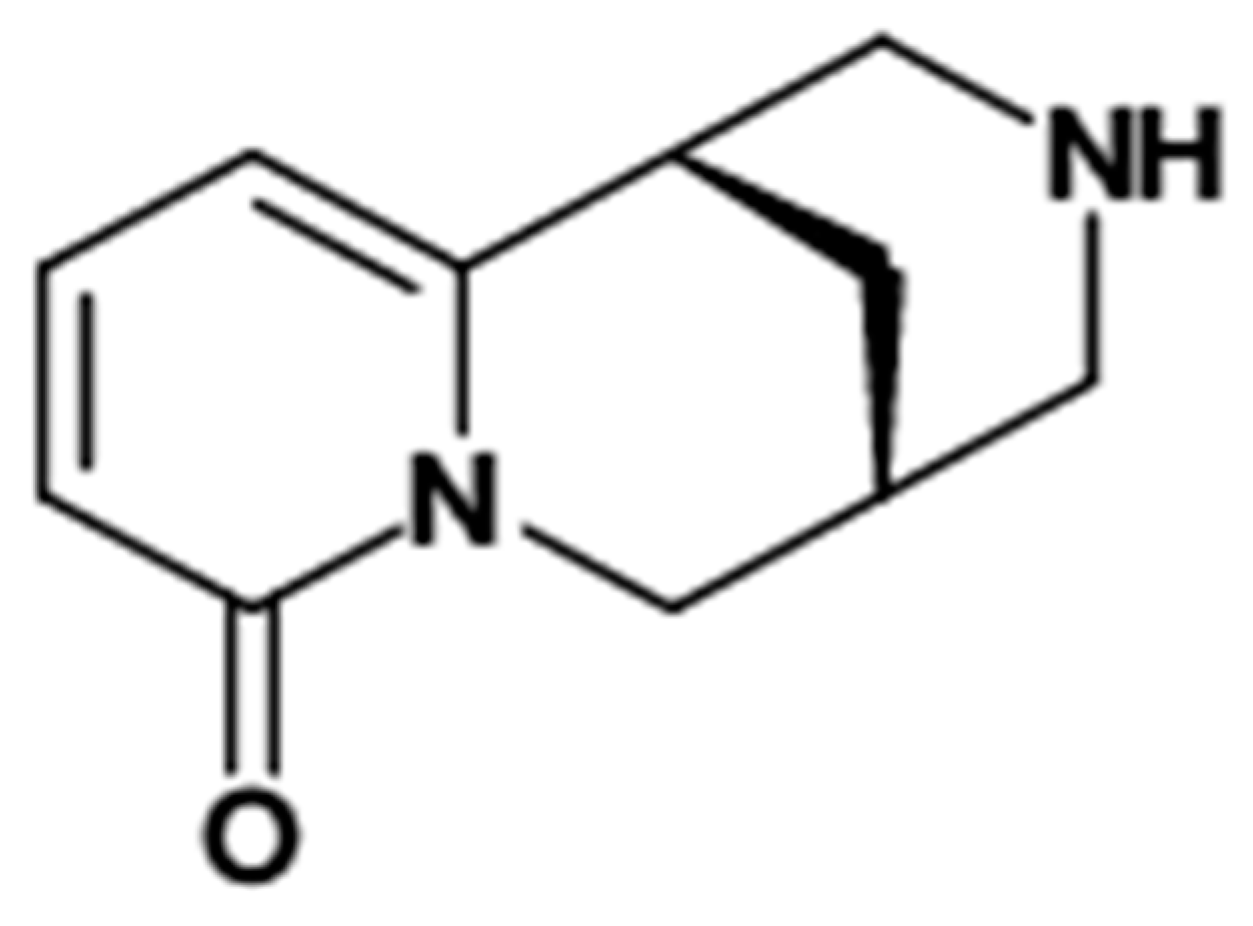
:
1. Introduction
2. Results and Discussion
2.1. Selection of Condition of Chromatographic Systems and Detection Techniques
2.1.1. Selection of Detection Techniques
2.1.2. Comparison of Chromatographic Systems
Analysis of Cytisine on Octadecyl Stationary Phase
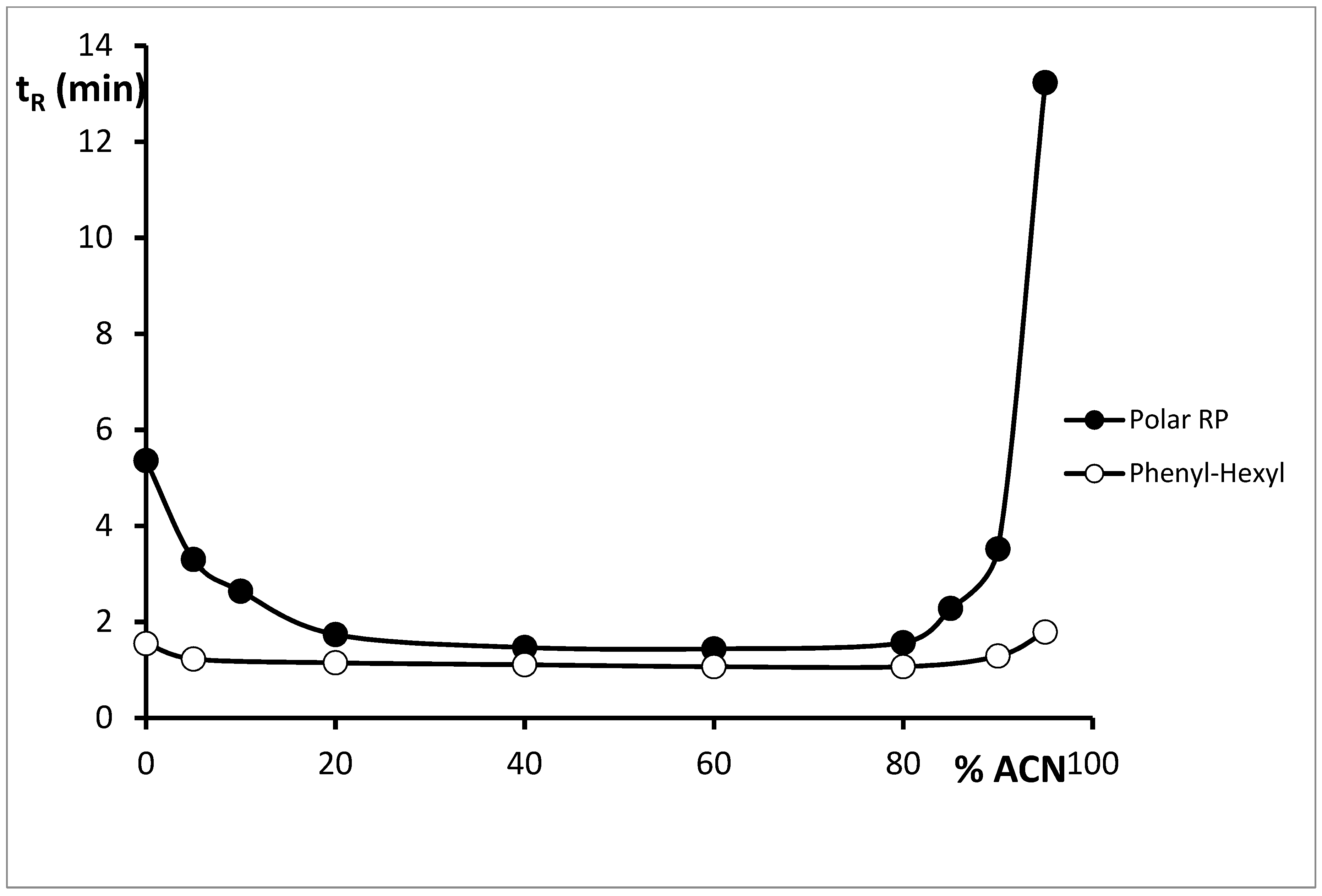
Analysis of Cytisine on Phenyl Stationary Phases
Analysis of Cytisine by HILIC
Analysis of Cytisine by Ion-Exchange Chromatography
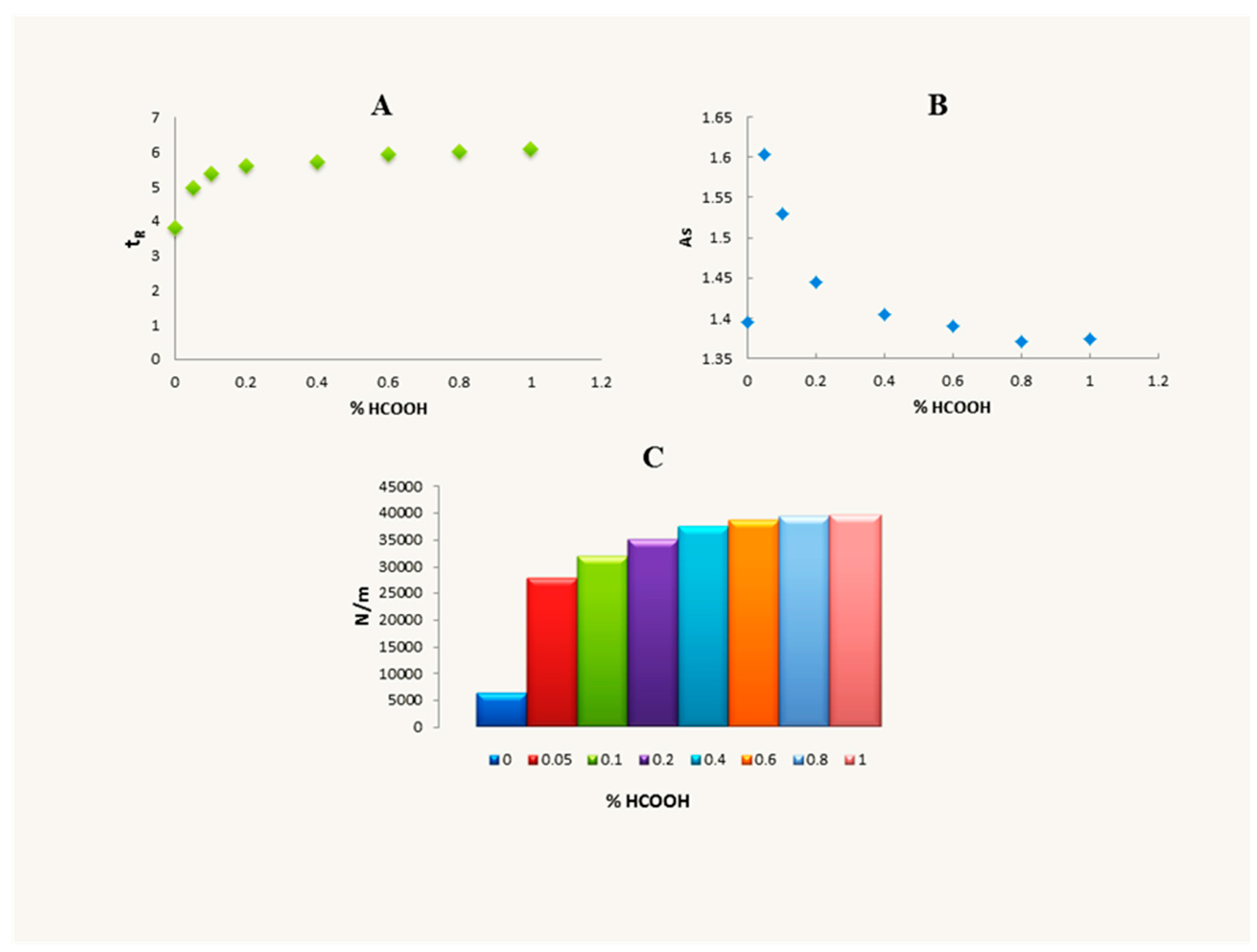
2.2. Optimisation of Sample Pre-Treatment Method
2.3. Analysis of Cytisine in Serum and Saliva Samples
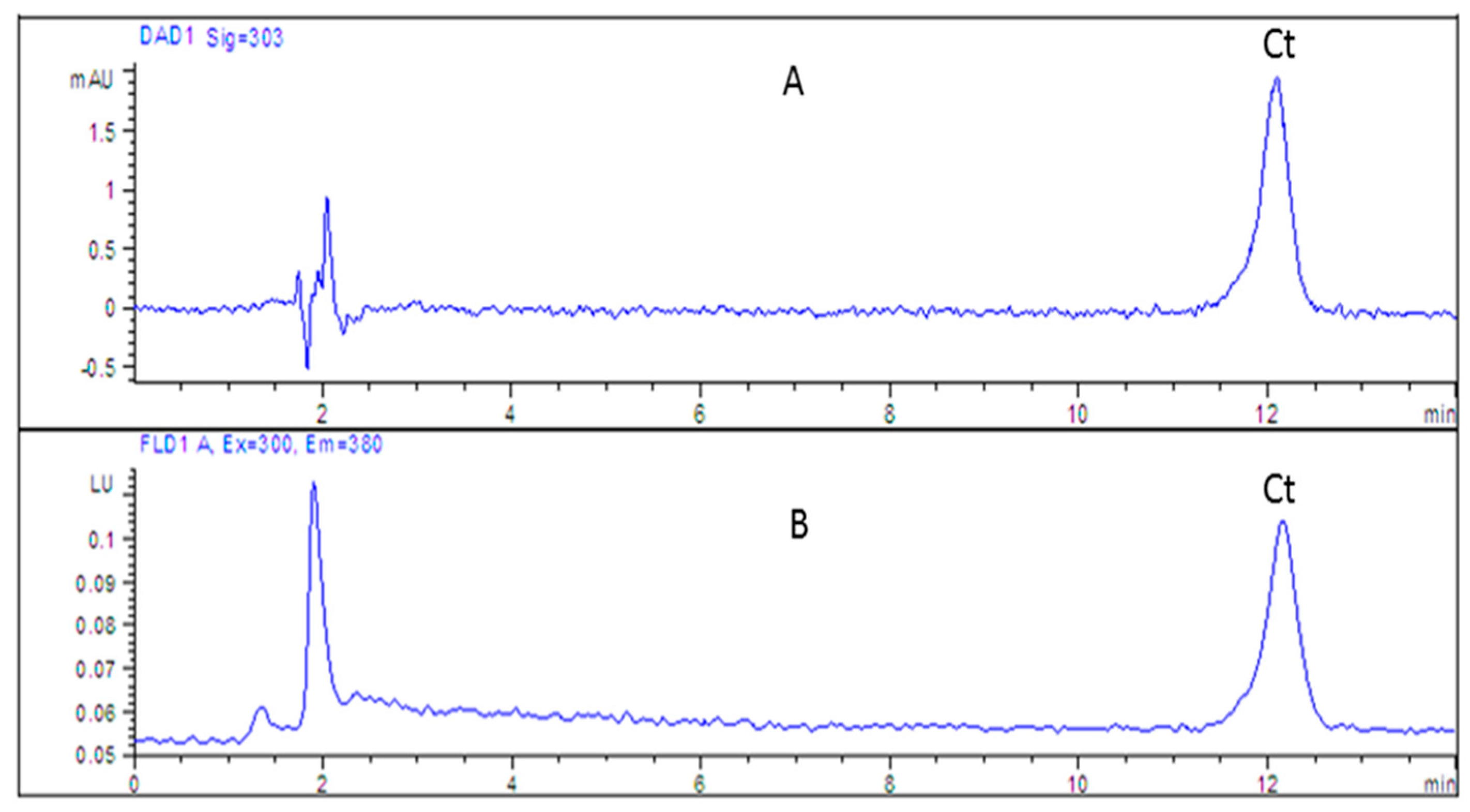
2.3.1. Analysis of Cytisine in Serum and Saliva Samples by HPLC-DAD
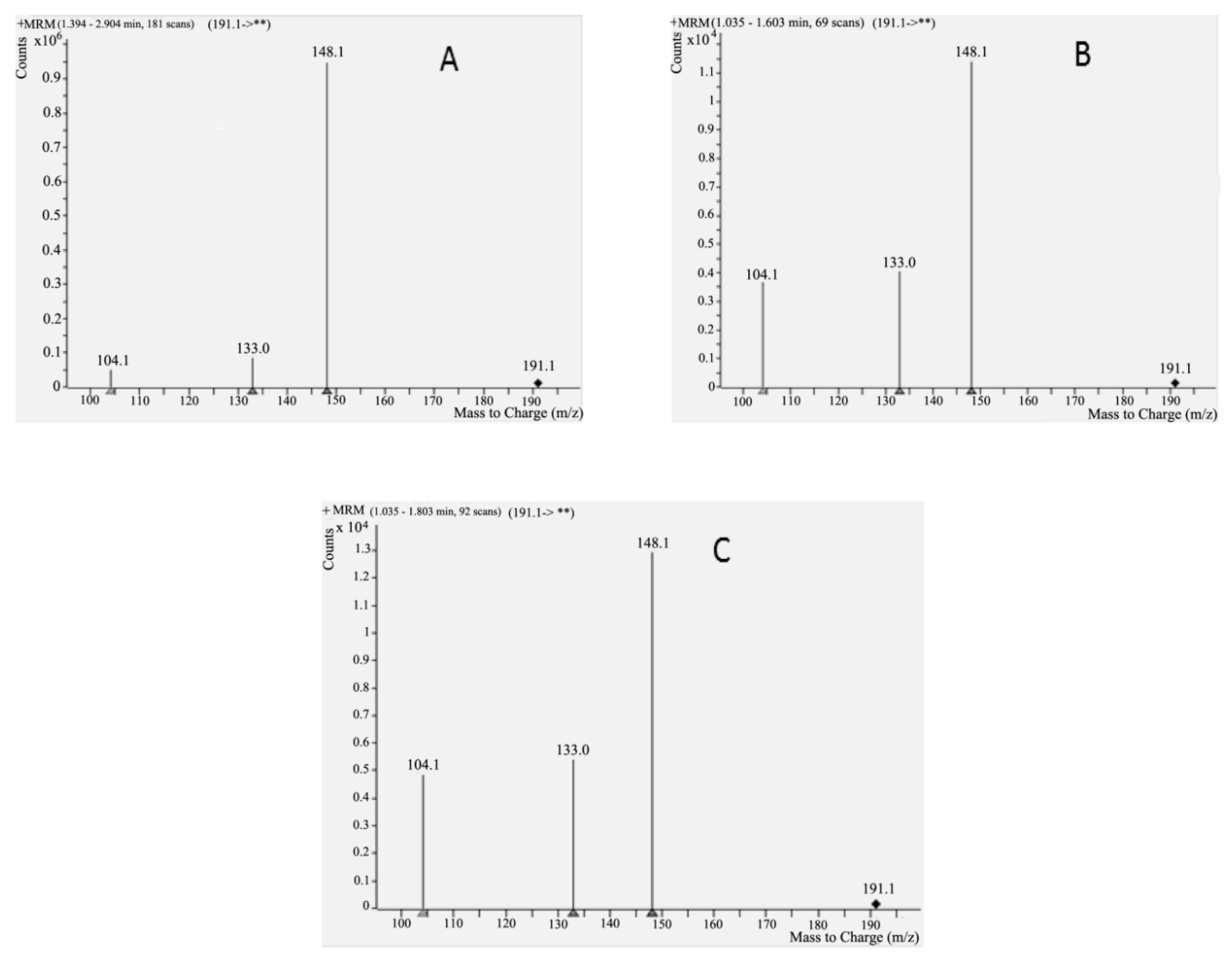
2.3.2. Analysis of Cytisine in Serum and Saliva Samples by HPLC-MS
2.4. Analysis of Cytisine in Pharmaceutical Formulation
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Apparatus and HPLC Conditions
3.2.1. HPLC-DAD Conditions
3.2.2. HPLC-FLD Conditions
3.2.3. HPLC-MS/MS Conditions
3.3. Serum and Saliva Samples Collection
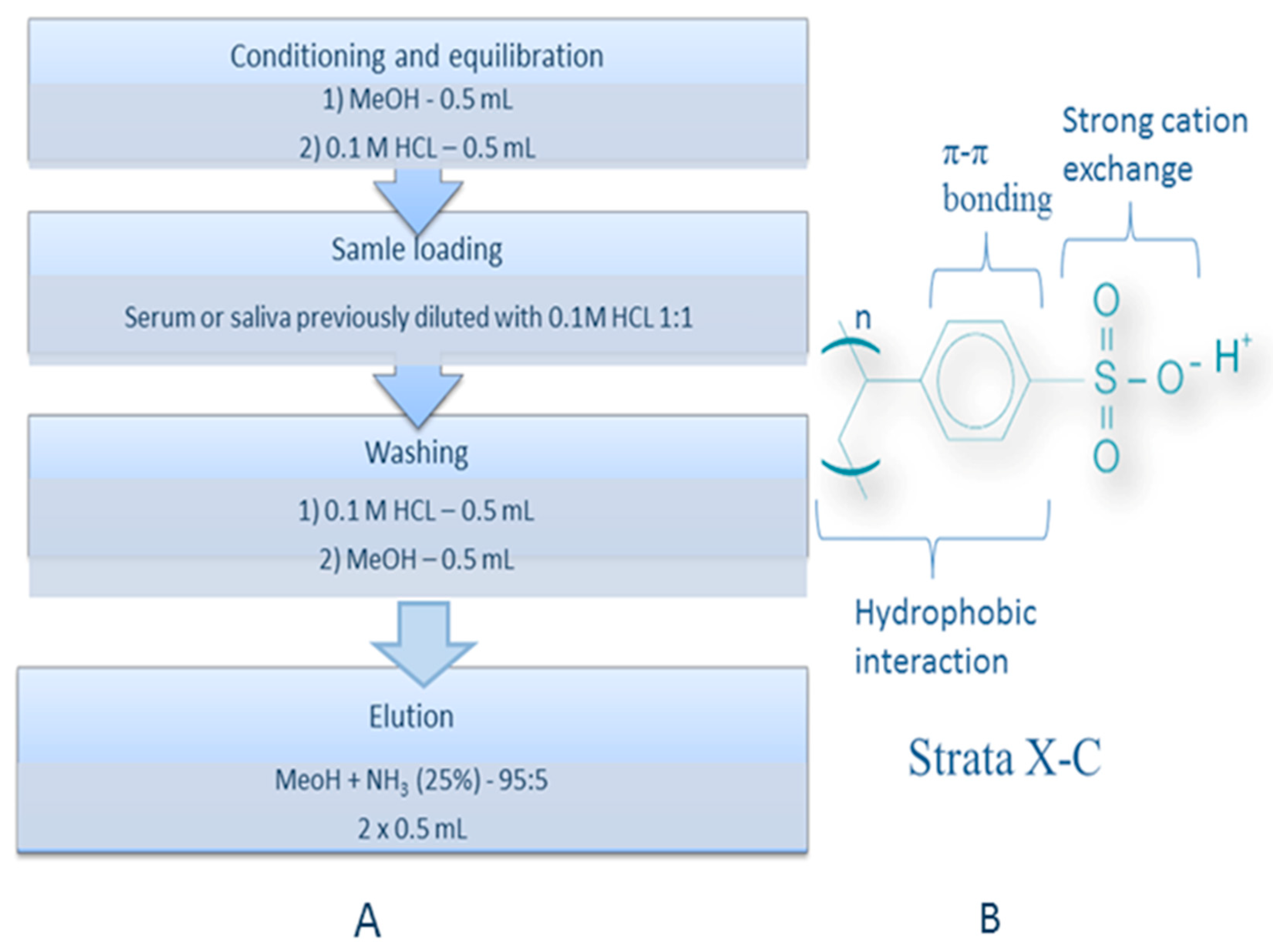
3.4. Extraction Procedure for Isolation of Investigated Drugs from sErum and Saliva
3.5. Preparation of Stock Solution and Working Solutions
3.6. Procedure for Analysis of Cytisine in Pharmaceutical Formulation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tutka, P.; Zatoński, W. Cytisine for the treatment of nicotine addiction: From a molecule to therapeutic efficacy. Pharmacol. Rep. 2006, 58, 777–798. [Google Scholar] [PubMed]
- Prochaska, J.J.; Das, S.; Benowitz, N.L. Cytisine, the world’s oldest smoking cessation aid. BMJ 2013, 347, f5198. [Google Scholar] [CrossRef] [PubMed]
- Tutka, P.; Vinnikov, D.; Courtney, R.C.; Benowitz, N.L. Cytisine for nicotine addiction treatment: A review of pharmacology, therapeutics and an update of clinical trial evidence for smoking cessation. Addiction 2019, (in press). [Google Scholar] [CrossRef] [PubMed]
- Rollema, H.; Hurst, R.S. The contribution of agonist and antagonist activities of α4β2* nAChR ligands to smoking cessation efficacy: A quantitative analysis of literature data. Psychopharmacology (Berl) 2018, 235, 2479–2505. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.; Bullen, C.; Barnes, J.; McRobbie, H.; Tutka, P.; Raw, M.; Etter, J.-F.; Siddiqi, K.; Courtney, R.J.; Castaldelli-Maia, J.M.; et al. Getting cytisine licensed for use world-wide: A call to action. Addiction 2016, 111, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.; Howe, C.; Glover, M.; McRobbie, H.; Barnes, J.; Nosa, V.; Parag, V.; Bassett, B.; Bullen, C. Cytisine versus nicotine for smoking cessation. N. Engl. J. Med. 2014, 371, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- West, R.; Zatonski, W.; Cedzynska, M.; Lewandowska, D.; Pazik, J.; Aveyard, P.; Stapleton, J. Placebo-Controlled Trial of Cytisine for Smoking Cessation. N. Engl. J. Med. 2011, 365, 1193–1200. [Google Scholar] [CrossRef]
- Vinnikov, D.; Brimkulov, N.; Burjubaeva, A. A Double-Blind, Randomised, Placebo-Controlled Trial of Cytisine for Smoking Cessation in Medium-Dependent Workers. J. Smok. Cessat. 2008, 3, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Hajek, P.; McRobbie, H.; Myers, K. Efficacy of cytisine in helping smokers quit: Systematic review and meta-analysis. Thorax 2013, 68, 1037–1042. [Google Scholar] [CrossRef]
- Leaviss, J.; Sullivan, W.; Ren, S.; Everson-Hock, E.; Stevenson, M.; Stevens, J.W.; Strong, M.; Cantrell, A. What is the clinical effectiveness and cost-effectiveness of cytisine compared with varenicline for smoking cessation? Health Technol. Assess. 2014, 18, 1–120. [Google Scholar] [CrossRef]
- Ebbert, J.O.; Wyatt, K.D.; Hays, J.T.; Klee, E.W.; Hurt, R.D. Varenicline for smoking cessation: Efficacy, safety, and treatment recommendations. Patient Prefer. Adher. 2010, 4, 355–362. [Google Scholar] [CrossRef]
- Etter, J.-F.; Lukas, R.J.; Benowitz, N.L.; West, R.; Dresler, C.M. Cytisine for smoking cessation: A research agenda. Drug Alcohol. Depend. 2008, 92, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, S.; Qian, D.; Qian, Y.; Duan, J.A. Comparative analysis of quinolizidine alkaloids from different parts of Sophora alopecuroides seeds by UPLC-MS/MS. J. Pharm. Biomed. Anal. 2012, 67–68, 16–27. [Google Scholar] [CrossRef]
- Ding, P.L.; Yu, Y.Q.; Chen, D.F. Determination of quinolizidine alkaloids in Sophora tonkinensis by HPCE. Phytochem. Anal. 2005, 16, 257–263. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Z.; He, L. An approach to develop binary chromatographic fingerprints of the total alkaloids from Caulophyllum robustum by high performance liquid chromatography/diode array detector and gas chromatography/mass spectrometry. J. Pharm. Biomed. Anal. 2007, 43, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, J.; Zhang, J.; Wei, S.; Ji, H.; Wu, X.; Mae, W.; Chen, J. Different processing methods change the oral toxicity induced by Sophora alopecuroides seeds and the contents of five main toxic alkaloids from the ethanol extracts determined by a validated UHPLC–MS/MS assay. Rev. Bras. Farmacogn. 2018, 28, 481–488. [Google Scholar] [CrossRef]
- Nirogi, R.; Kandikere, V.; Bhyrapuneni, G.; Saralaya, R.; Muddana, N.; Ajjala, D.R. Rat thalamic α4β2 neuronal nicotinic acetylcholine receptor occupancy assay using LC–MS/MS. J. Pharmacol. Tox. Met. 2012, 65, 136–141. [Google Scholar] [CrossRef]
- Reavill, C.; Walther, B.; Stolerman, I.P.; Testa, B. Behavioural and pharmacokinetic studies on nicotine, cytisine and lobeline. Neuropharmacology 1990, 29, 619–624. [Google Scholar] [CrossRef]
- Astroug, H.; Simeonova, R.; Kassabova, L.V.; Danchev, N.; Svinarov, D. Pharmacokinetics of cytisine after single intravenous and oral administration in rabbits. Interdiscip. Toxicol. 2010, 3, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, J.; Gunther, J.; Henle, T.; Dressler, J. Simultaneous determination of thirteen plant alkaloids in a human specimen by SPE and HPLC. J. Sep. Sci. 2008, 31, 2410–2416. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.; Peters, F.T.; Kraemer, T.; Maurer, H.H. Detection and validated quantification of toxic alkaloids in human blood plasma-comparison of LC-APCI-MS with LC-ESI-MS/MS. J. Mass Spectrom. 2007, 42, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Borucka, B.; Sawicka, R.; Grabowski, T.; Świerczewska, A.; Raszewski, W. New sensitive LC-MS method for cytisine determination in human plasma and urine. Validation and quality assurance during study. In Proceedings of the I Conference of Polish Mass Spectrometry Society, Puławy, Poland, 16–18 April 2008; p. 52. [Google Scholar]
- Jeong, S.H.; Newcombe, D.; Sheridan, J.; Tingle, M. Pharmacokinetics of cytisine, an α4β2 nicotinic receptor partial agonist, in healthy smokers following a single dose. Drug Test. Anal. 2015, 7, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Carlier, J.; Guitton, J.; Romeuf, L.; Bévalot, F.; Boyer, B.; Fanton, L.; Gaillard, Y. Screening approach by ultra-high performance liquid chromatography-tandem mass spectrometry for the blood quantification of thirty-four toxic principles of plant origin. Application to forensic toxicology. J. Chromatogr. B 2015, 975, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Ching, C.K.; Chan, A.Y.W.; Mak, T.W.L. Simultaneous detection of 22 toxic plant alkaloids (aconitum alkaloids, solanaceous tropane alkaloids, sophora alkaloids, strychnos alkaloids and colchicine) in human urine and herbal samples using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2013, 942–943, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Musshoff, F.; Madea, B. Fatal cytisine intoxication and analysis of biological samples with LC-MS/MS. Forensic Sci. Int. 2009, 186, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Richards, H.G.; Stephens, A. A fatal case of laburnum seed poisoing. Med. Sci. Law. 1970, 10, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Pieńko, T.; Grudzień, M.; Taciak, P.P.; Mazurek, A.P. Cytisine basicity, solvatation, logP, and logD theoretical determination as tool for bioavailability prediction. J. Mol. Graph. Model. 2016, 63, 15–21. [Google Scholar] [CrossRef]
- Krystkowiak, E.; Przybył, A.K.; Bayda-Smykaj, M.; Koput, J.; Maciejewski, A. Spectral and photophysical properties of cytisine in acetonitrile—Theory and experiment. Spectrochim. Acta A 2018, 203, 375–382. [Google Scholar] [CrossRef]
- Jeong, S.H.; Sheridan, J.; Newcombe, D.; Tingle, M. Plasma concentrations of cytisine, a commercially available plant-based alkaloid, in healthy adult smokers taking recommended doses for smoking cessation. Xenobiotica 2018, 48, 1245–1248. [Google Scholar] [CrossRef]
- Jeong, S.H.; Sheridan, J.; Bullen, C.; Newcombe, D.; Walker, N.; Tingle, M. Ascending single dose pharmacokinetics of cytisine in healthy adult smokers. Xenobiotica 2018. [Google Scholar] [CrossRef]
- Petruczynik, A.; Waksmundzka-Hajnos, M. Effect of Chromatographic Conditions on the Separation of Selected Alkaloids on Phenyl Stationary Phase by an HPLC Method. J. Liquid Chromatogr. 2006, 29, 2807–2822. [Google Scholar] [CrossRef]
- Petruczynik, A. Effect of Ionic Liquid Additives to Mobile Phase on Separation and System Efficiency for HPLC of Selected Alkaloids on Different Stationary Phases. J. Chromatogr. Sci. 2012, 50, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Column | Functional Group | Length (mm) | (Inner Diameter) I.D. (mm) | Endcapped | Particle Size (μm) | Pore Size (Å) | Surface Area (m2/g) | Carbon Load (%) | Recommended pH Range |
|---|---|---|---|---|---|---|---|---|---|
| Synergi Polar RP | Ether-linked phenyl | 150 | 4.6 | Proprietary (polar group) | 4 | 80 | 475 | 11 | 1.5–7.0 |
| Synergi HST Polar RP | Ether-linked phenyl | 100 | 2 | Proprietary (polar group) | 2.5 | 100 | 475 | 11 | 1.5–7.0 |
| CSH Phenyl-Hexyl | Phenyl-Hexyl | 150 | 4.6 | Proprietary | 5 | 130 | 185 | 15 | 1.0–11.0 |
| Synergi Hydro-RP | Octadecyl (C18) | 150 | 4.6 | Proprietary (polar group) | 4 | 80 | 475 | 19 | 1.5–7.5 |
| ACE HILIC-A | Proprietary SIL | 150 | 4.6 | NO | 5 | 100 | 300 | - | 2.0–7.0 |
| ACE HILIC-B | Proprietary Aminopropyl | 150 | 4.6 | NO | 5 | 100 | 300 | 4 | 2.0–7.0 |
| ACE HILIC-C | Proprietary Polyhydroxy | 150 | 4.6 | NO | 5 | 100 | 300 | 7 | 2.0–7.0 |
| Luna SCX | Benzene Sulfonic Acid | 150 | 4.6 | NO | 5 | 100 | 400 | 0.55 Sulphur Load | 2.0–7.0 |
| Column | Mobile Phase | tR | As | N/m |
|---|---|---|---|---|
| Hydro RP | 5% MeCN + H2O + 0.1% HCOOH | 1.76 | * | * |
| 5% MeOH + H2O + 0.1% HCOOH | 1.85 | * | * | |
| H2O + 0.1% HCOOH | 1.92 | * | * | |
| 5% MeOH + 20% acetate buffer at pH 3.5 H2O + 0.025ML−1 DEA | 1.91 | * | * | |
| 20% acetate buffer at pH 3.5H2O + 0.025ML−1 DEA | 1.56 | * | * | |
| 5% MeCN + H2O + 0.025ML−1 NaBF4 | 2.98 | * | * | |
| 10% MeCN + H2O + 0.025ML−1 ILBF4 | 2.96 | * | 11,120 | |
| H2O + 0.025ML−1 IL BF4 | 3.46 | * | 12,740 | |
| Phenyl-Hexyl | 5% MeCN + H2O + 0.1% HCOOH | 1.23 | * | * |
| 5% MeOH + H2O + 0.1% HCOOH | 1.38 | * | * | |
| H2O + 0.1% HCOOH | 1.55 | * | * | |
| 5% MeCN + 20% acetate buffer at pH 3.5 H2O + 0.025ML−1 DEA | 3.49 | 0.73 | 13,200 | |
| 5% MeOH + 20% acetate buffer at pH 3.5 H2O + 0.025ML−1 DEA | 3.64 | 0.60 | * | |
| 20% acetate buffer at pH 3.5H2O + 0.025ML−1 DEA | 5.29 | 0.64 | 10,070 | |
| H2O + 0.025ML−1 NaCl | * | |||
| H2O + 0.025ML−1 IL Cl | 2.45 | 1.04 | 12,140 | |
| H2O + 0.025ML−1 NaBF4 | 6.69 | 1.60 | 13,390 | |
| H2O + 0.025ML−1 IL BF4 | 3.25 | 1.08 | 12,240 | |
| Polar RP | 5% MeCN + H2O + 0.1% HCOOH | 3.30 | * | * |
| 5% MeOH + H2O + 0.1% HCOOH | 4.10 | * | * | |
| H2O + 0.1% HCOOH | 5.31 | 1.50 | 37,260 | |
| 5% MeCN + 20% acetate buffer at pH 3.5 H2O + 0.025ML−1 DEA | 3.49 | 1.31 | 31,830 | |
| 5% MeOH + 20% acetate buffer at pH 3.5 H2O + 0.025ML−1 DEA | 4.49 | 1.33 | 23,750 | |
| 20% acetate buffer at pH 3.5 + H2O + 0.025ML−1 DEA | 5.90 | 1.47 | 21,030 | |
| 5% MeCN + H2O + 0.025ML−1 NaH2PO4 | 3.76 | 1.22 | 41,870 | |
| H2O + 0.025ML−1 IL H2PO4 | 2.66 | 1.97 | 16,610 | |
| 5% MeCN + H2O + 0.025ML−1 NaMeSO3 | 4.07 | 1.20 | 44,320 | |
| H2O + 0.025ML−1 IL MeSO3 | 2.10 | 1.93 | 13,920 | |
| 5% MeCN + H2O + 0.025ML−1 NaCl | 3.91 | 1.19 | 44,570 | |
| H2O + 0.025ML−1 IL Cl | 5.91 | 0.81 | 22,400 | |
| 5% MeCN + H2O + 0.025ML−1 NaBF4 | 5.19 | 1.11 | 55,120 | |
| H2O + 0.025ML−1 IL BF4 | 3.59 | 0.66 | 7250 | |
| 5% MeCN + H2O + 0.025ML−1 NaClO4 | 6.33 | 1.16 | 56,230 | |
| H2O + 0.025ML−1 IL ClO4 | 5.58 | 1.15 | 45,580 | |
| 5% MeCN + H2O + 0.025ML−1 NaPF6 | 8.95 | 1.04 | 69,470 | |
| H2O + 0.025ML−1 IL PF6 | 3.56 | 1.02 | 28,850 | |
| HILIC A | 80% MeCN + H2O + 0.1 ML−1 HCOONH4 | 3.91 | * | * |
| 80% MeCN + formate buffer at pH 4.0 | 6.82 | 1.08 | 56,940 | |
| HILIC B | 80% MeCN + H2O + 0.1 ML−1 HCOONH4 | 2.96 | 0.80 | 10,330 |
| 80% MeCN + formate buffer at pH 4.0 | 9.61 | 1.51 | 4680 | |
| HILIC N | 80% MeCN + H2O + 0.1 ML−1 HCOONH4 | 4.38 | 1.25 | 33,290 |
| 80% MeCN + formate buffer at pH 4.0 | 4.74 | 1.11 | 40,890 | |
| SCX | 15% MeCN + phosphoric buffer at pH 2.5 | 5.13 | 0.96 | 48,670 |
| 15% MeCN + 25 mM formate buffer at pH 4.0 | 48.62 | 1.04 | 75,520 | |
| 15% MeCN + 50 mM formate buffer at pH 4.0 | 25.82 | 0.97 | 66,360 | |
| 15% MeCN + 75 mM formate buffer at pH 4.0 | 18.16 | 0.97 | 61,720 | |
| 15% MeCN + 100 mM formate buffer at pH 4.0 | 14.44 | 0.97 | 58,780 | |
| 15% MeCN + 150 mM formate buffer at pH 4.0 | 10.74 | 0.97 | 53,810 | |
| 15% MeCN + 200 mM formate buffer at pH 4.0 | 8.91 | 0.98 | 51,200 |
| Precursor Ion | Fragmentor | Product Ion | Collision Energy |
|---|---|---|---|
| 191.12 | 148 | 148.1 | 20 |
| 133.0 | 36 | ||
| 104.1 | 60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróblewski, K.; Petruczynik, A.; Tuzimski, T.; Przygodzka, D.; Buszewicz, G.; Kołodziejczyk, P.; Tutka, P. Comparison of Various Chromatographic Systems for Analysis of Cytisine in Human Serum, Saliva and Pharmaceutical Formulation by HPLC with Diode Array, Fluorescence or Mass Spectrometry Detection. Molecules 2019, 24, 2580. https://doi.org/10.3390/molecules24142580
Wróblewski K, Petruczynik A, Tuzimski T, Przygodzka D, Buszewicz G, Kołodziejczyk P, Tutka P. Comparison of Various Chromatographic Systems for Analysis of Cytisine in Human Serum, Saliva and Pharmaceutical Formulation by HPLC with Diode Array, Fluorescence or Mass Spectrometry Detection. Molecules. 2019; 24(14):2580. https://doi.org/10.3390/molecules24142580
Chicago/Turabian StyleWróblewski, Karol, Anna Petruczynik, Tomasz Tuzimski, Dominika Przygodzka, Grzegorz Buszewicz, Patrycjusz Kołodziejczyk, and Piotr Tutka. 2019. "Comparison of Various Chromatographic Systems for Analysis of Cytisine in Human Serum, Saliva and Pharmaceutical Formulation by HPLC with Diode Array, Fluorescence or Mass Spectrometry Detection" Molecules 24, no. 14: 2580. https://doi.org/10.3390/molecules24142580
APA StyleWróblewski, K., Petruczynik, A., Tuzimski, T., Przygodzka, D., Buszewicz, G., Kołodziejczyk, P., & Tutka, P. (2019). Comparison of Various Chromatographic Systems for Analysis of Cytisine in Human Serum, Saliva and Pharmaceutical Formulation by HPLC with Diode Array, Fluorescence or Mass Spectrometry Detection. Molecules, 24(14), 2580. https://doi.org/10.3390/molecules24142580