
Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry



Abstract
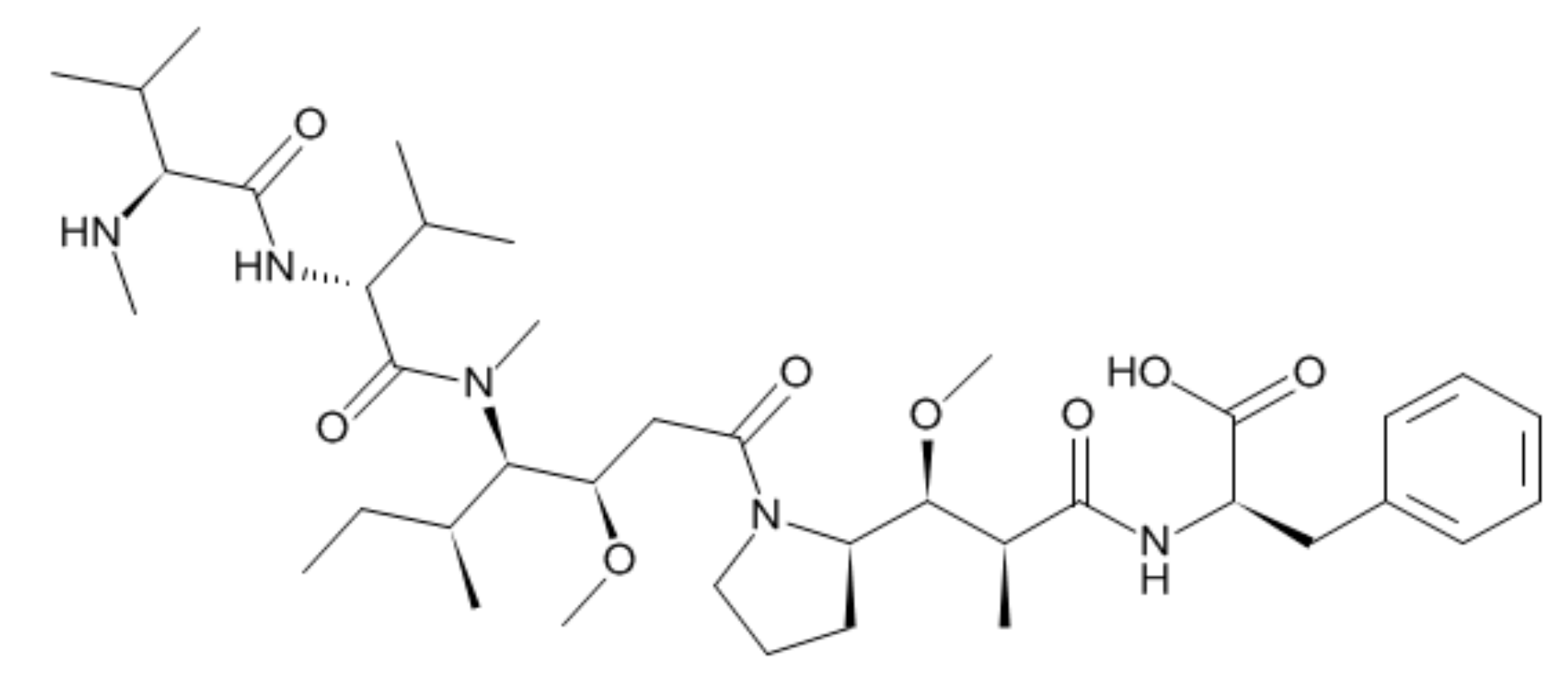
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Stock Solution, Calibration Standard (STD), and Quality Control (QC) Samples
2.3. Sample Preparation (Plasma Sample)
2.4. LC-TOF-MS/MS Conditions
2.5. Method Qualification
2.6. Software
2.7. Application for a Pharmacokinetic Study in Rat
2.8. Sample Preparation—In Vitro/In Vivo Metabolite Identification (Met ID)
3. Results and Discussion
3.1. Method Development and Qualification
3.1.1. Sample Preparation and Optimization of LC-TOF-MS/MS Parameters
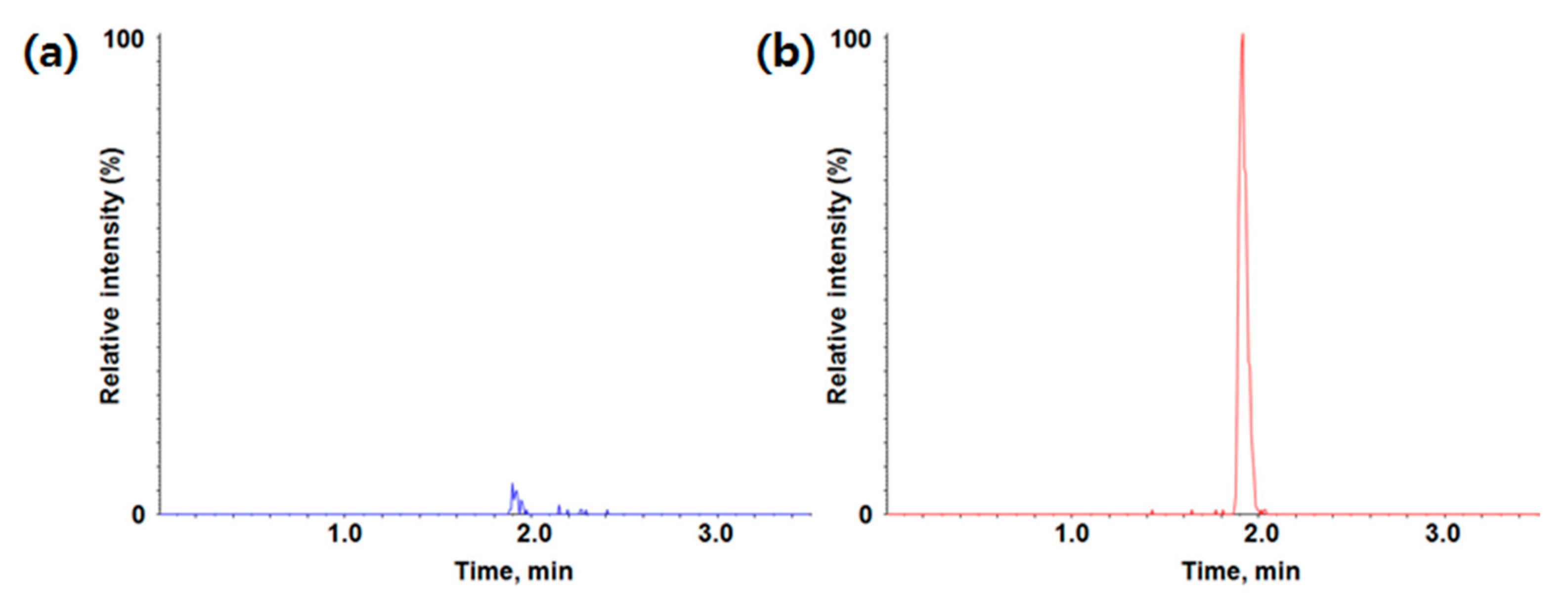
3.1.2. Method Qualification
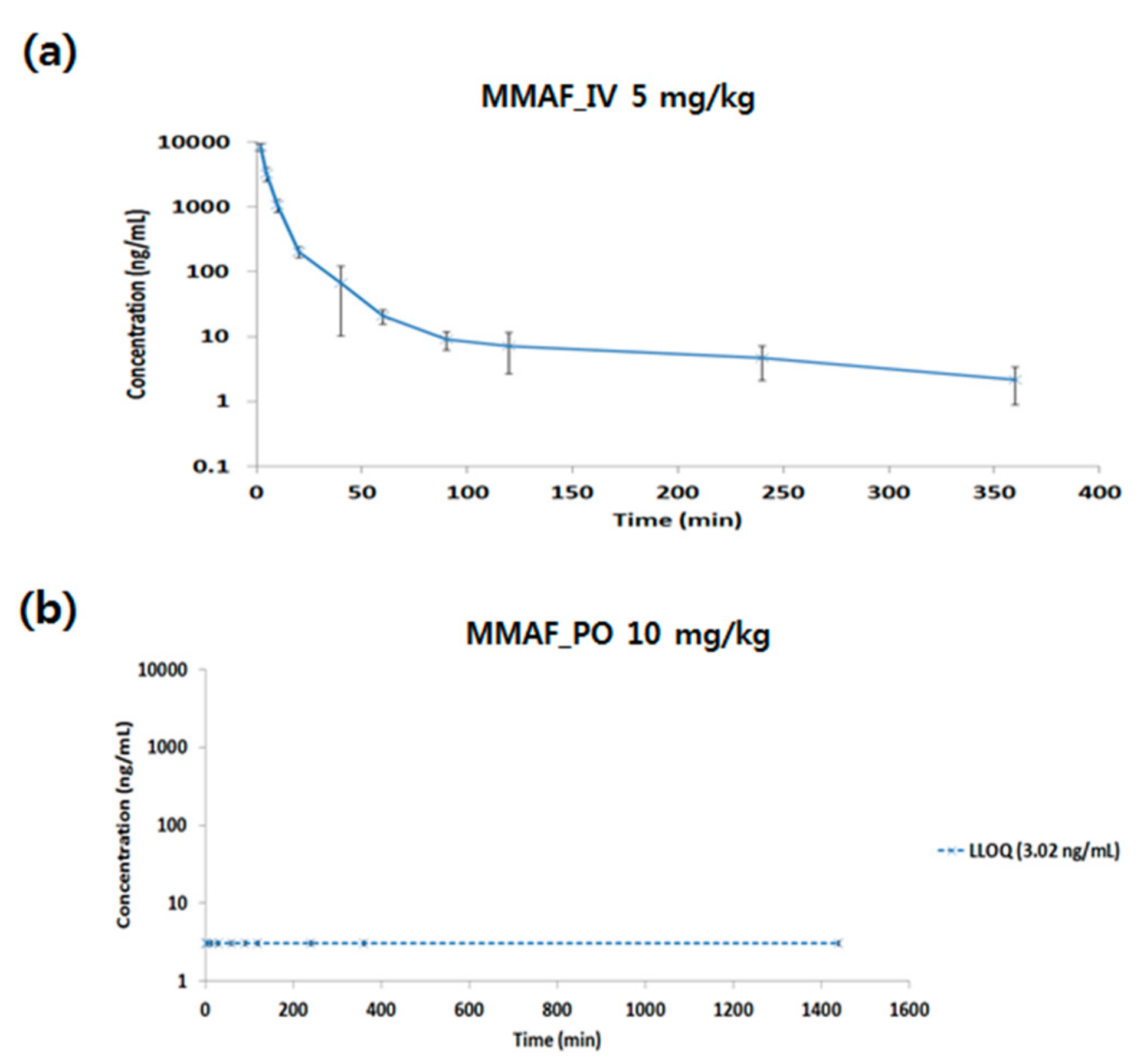
3.2. Application for Pharmacokinetic Study
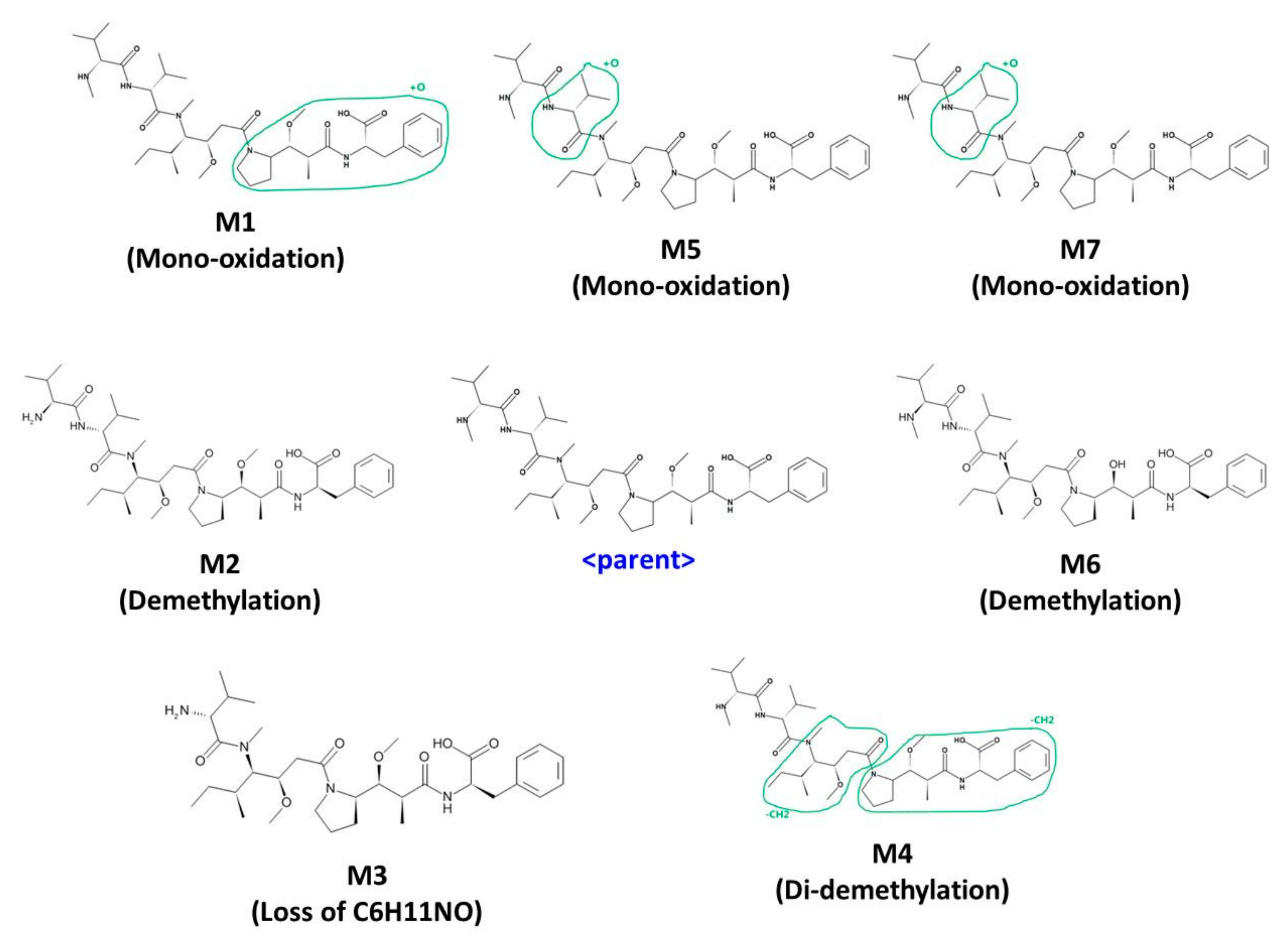
3.3. In Vitro and In Vivo Metabolite Profiling for MMAF
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody-drug conjugates. Nat. Rev. Drug. Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Reichert, J.M. Antibody-drug conjugates: Present and future. MAbs 2014, 6, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Kizlik-Masson, C.; Pelegrin, A.; Watier, H.; Viaud-Massuard, M.C.; Joubert, N. Antibody-drug conjugates: Design and development for therapy and imaging in and beyond cancer, LabEx MAbImprove industrial workshop, July 27-28, 2017, Tours, France. MAbs 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control Rel. 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Goad, M.E.; Dougher, M.M.; Boghaert, E.R.; Kunz, A.; Hamann, P.R.; Damle, N.K. Potent and specific antitumor efficacy of CMC-544, a CD22-targeted immunoconjugate of calicheamicin, against systemically disseminated B-cell lymphoma. Clin. Cancer Res. 2004, 10, 8620–8629. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.F.; Klussman, K.; Thompson, J.D.; Chen, J.H.; Francisco, L.V.; Risdon, G.; Chace, D.F.; Siegall, C.B.; Francisco, J.A. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res. 2002, 62, 3736–3742. [Google Scholar]
- Ojima, I.; Geng, X.D.; Wu, X.Y.; Qu, C.X.; Borella, C.P.; Xie, H.S.; Wilhelm, S.D.; Leece, B.A.; Bartle, L.M.; Goldmacher, V.S.; et al. Tumor-specific novel taxoid-monoclonal antibody conjugates. J. Med. Chem. 2002, 45, 5620–5623. [Google Scholar] [CrossRef]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Structure-activity studies with chiral isomers and with segments of the antimitotic marine peptide dolastatin 10. Biochem. Pharmacol. 1990, 40, 1859–1864. [Google Scholar] [CrossRef]
- Brase, S.; Glaser, F.; Kramer, C.S.; Lindner, S.; Linsenmeier, A.M.; Masters, K.S.; Meister, A.C.; Ruff, B.M.; Zhong, S. Progress in the chemistry of organic natural products. The chemistry of mycotoxins. Prog. Chem. Org. Nat. Prod. 2013, 97, 1–300. [Google Scholar]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef]
- Fennell, B.J.; Carolan, S.; Pettit, G.R.; Bell, A. Effects of the antimitotic natural product dolastatin 10, and related peptides, on the human malarial parasite Plasmodium falciparum. J. Antimicrob. Chemother. 2003, 51, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Pettit, R.K.; Pettit, G.R.; Hazen, K.C. Specific activities of dolastatin 10 and peptide derivatives against Cryptococcus neoformans. Antimicrob. Agents Chemother. 1998, 42, 2961–2965. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.Q.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.J.; Li, Y.Y.; Liu, X.Y.; Lu, X.L.; Cao, X.; Jiao, B.H. Marine Antibody-Drug Conjugates: Design Strategies and Research Progress. Mar Drugs 2017, 15. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjugate Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Sedlik, C.; Heitzmann, A.; Viel, S.; Sarkouh, R.A.; Batisse, C.; Schmidt, F.; De la Rochere, P.; Amzallag, N.; Osinaga, E.; Oppezzo, P.; et al. Effective antitumor therapy based on a novel antibody-drug conjugate targeting the Tn carbohydrate antigen. Oncoimmunology 2016, 5. [Google Scholar] [CrossRef]
- Walker, D.K. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Brit. J. Clin. Pharmacol. 2004, 58, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Setchell, K.D.R.; Brown, N.M.; Desai, P.; Zimmer-Nechemias, L.; Wolfe, B.E.; Brashear, W.T.; Kirschner, A.S.; Cassidy, A.; Heubi, J.E. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J. Nutr. 2001, 131, 1362s–1375s. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Lue, J.K.; Sawas, A.; Amengual, J.E.; Deng, C.C.; Kalac, M.; Falchi, L.; Marchi, E.; Turenne, I.; Lichtenstein, R.; et al. Brentuximab vedotin plus bendamustine in relapsed or refractory Hodgkin’s lymphoma: an international, multicentre, single-arm, phase 1-2 trial. Lancet Oncol. 2018, 19, 257–266. [Google Scholar] [CrossRef]
- Cole, P.D.; Metzger, M.; Drachtman, R.A.; Horton, T.M.; Liu, X.W.; Ahern, C.H.; Minard, C.; Fox, E.; Blaney, S.; Weigel, B.; et al. Phase 1 trial of brentuximab vedotin in combination with gemcitabine for pediatric and young adult patients with relapsed or refractory Hodgkin lymphoma, a Children’s Oncology Group report. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Akaiwa, M.; Martin, T.; Mendelsohn, B.A. Synthesis and Evaluation of Linear and Macrocyclic Dolastatin 10 Analogues Containing Pyrrolidine Ring Modifications. ACS Omega 2018, 3, 5212–5221. [Google Scholar] [CrossRef]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef]
- Garteiz, D.A.; Madden, T.; Beck, D.E.; Huie, W.R.; McManus, K.T.; Abbruzzese, J.L.; Chen, W.; Newman, R.A. Quantitation of dolastatin-10 using HPLC/electrospray ionization mass spectrometry: application in a phase I clinical trial. Cancer Chemother. Pharm. 1998, 41, 299–306. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, Y.; Lou, Y.; Jiang, H.; Wang, X.; Chai, X.; Zeng, S. Metabolism of ebracteolata compound B studied in vitro with human liver microsomes, HepG2 cells, and recombinant human enzymes. Drug Metab. Dispos. 2010, 38, 2157–2165. [Google Scholar] [CrossRef]
- Vikingsson, S.; Wohlfarth, A.; Andersson, M.; Green, H.; Roman, M.; Josefsson, M.; Kugelberg, F.C.; Kronstrand, R. Identifying Metabolites of Meclonazepam by High-Resolution Mass Spectrometry Using Human Liver Microsomes, Hepatocytes, a Mouse Model, and Authentic Urine Samples. AAPS J. 2017, 19, 736–742. [Google Scholar] [CrossRef]
- Watanabe, S.; Vikingsson, S.; Roman, M.; Green, H.; Kronstrand, R.; Wohlfarth, A. In Vitro and In Vivo Metabolite Identification Studies for the New Synthetic Opioids Acetylfentanyl, Acrylfentanyl, Furanylfentanyl, and 4-Fluoro-Isobutyrylfentanyl. AAPS J. 2017, 19, 1102–1122. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.L.; Wu, M.J.; Chen, X.Z.; Ma, H.L.; Ding, L.Q.; Qiu, F.; Pan, Q.; Zhang, D.Q. Metabolic profiling of nuciferine in rat urine, plasma, bile and feces after oral administration using ultra-high performance liquid chromatography-diode array detection-quadrupole time-of-flight mass spectrometry. J. Pharm. Biomed. 2017, 140, 71–80. [Google Scholar] [CrossRef]
- Erickson, H.K.; Lambert, J.M. ADME of antibody-maytansinoid conjugates. AAPS J. 2012, 14, 799–805. [Google Scholar] [CrossRef]
- Hop, C.E.; Wang, Z.; Chen, Q.; Kwei, G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J. Pharm. Sci. 1998, 87, 901–903. [Google Scholar] [CrossRef]
- Hamilton, R.A.; Garnett, W.R.; Kline, B.J. Determination of mean valproic acid serum level by assay of a single pooled sample. Clin. Pharmacol. Ther. 1981, 29, 408–413. [Google Scholar] [CrossRef]
- Ramagiri, S.; Garofolo, F. Large molecule bioanalysis using Q-TOF without predigestion and its data processing challenges. Bioanalysis 2012, 529–540. [Google Scholar] [CrossRef]
- Shin, S.H.; Park, M.H.; Byeon, J.J.; Lee, B.I.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Pharmaceutics 2018, 10. [Google Scholar] [CrossRef]
- Lee, B.I.; Park, M.H.; Heo, S.C.; Park, Y.; Shin, S.H.; Byeon, J.J.; Kim, J.H.; Shin, Y.G. Quantification and application of a liquid chromatography-tandem mass spectrometric method for the determination of WKYMVm peptide in rat using solid-phase extraction. Biomed. Chromatogr. 2018, 32. [Google Scholar] [CrossRef]
- Lee, B.I.; Park, M.H.; Shin, S.H.; Byeon, J.J.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, Y.Y.; Cho, K.H.; La, S.; Lee, H.J.; Yim, D.S.; Ban, S.; Park, M.Y.; Kim, Y.C.; Kim, Y.G.; et al. Validation of a liquid chromatography-triple quadrupole mass spectrometric method for the determination of 5-nitro-5’-hydroxy-indirubin-3’-oxime (AGM-130) in human plasma and its application to microdose clinical trial. Biomed. Chromatogr. 2016, 30, 323–329. [Google Scholar] [CrossRef]
- Ren, L.J.; Wu, H.J.; Sun, L.H.; Xu, X.; Mo, L.Y.; Zhang, L.; Zhang, J.Y.; Wu, C.Y. A sensitive LC-MS/MS method for simultaneous determination of cabozantinib and its metabolite cabozantinib N-oxide in rat plasma and its application in a pharmacokinetic study. Biomed. Chromatogr. 2018, 32, e4227. [Google Scholar] [CrossRef]
- Zhang, M.; Eismin, R.; Kenttamaa, H.; Xiong, H.; Wu, Y.; Burdette, D.; Urbanek, R. Identification of 2-aminothiazolobenzazepine metabolites in human, rat, dog, and monkey microsomes by ion-molecule reactions in linear quadrupole ion trap mass spectrometry. Drug Metab. Dispos. 2015, 43, 358–366. [Google Scholar] [CrossRef]
- Amer, S.M.; Kadi, A.A.; Darwish, H.W.; Attwa, M.W. Identification and characterization of in vitro phase I and reactive metabolites of masitinib using a LC-MS/MS method: bioactivation pathway elucidation. RSC Adv. 2017, 7, 4479–4491. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantification of MMAF | |
|---|---|
| Time (min) | Mobile phase B (%) |
| 0 | 10 |
| 0.5 | 10 |
| 1.3 | 95 |
| 1.9 | 95 |
| 2.0 | 10 |
| 3.5 | 10 |
| Metabolite profiling of MMAF | |
| Time (min) | Mobile phase B (%) |
| 0 | 5 |
| 2 | 5 |
| 28 | 33 |
| 29 | 33 |
| 34 | 95 |
| 38 | 95 |
| 38.1 | 5 |
| 45 | 5 |
| Low QC (165.46 ng/mL) | High QC (1820 ng/mL) | |
|---|---|---|
| Mean concentration (ng/mL) | 163.94 | 1832.78 |
| Accuracy % | 99.08 | 100.70 |
| % CV | 2.52 | 2.83 |
| n | 10 | 10 |
| Low QC (165.46 ng/mL) | |||||
|---|---|---|---|---|---|
| Species | Mouse | Rat | Dog | Monkey | Human |
| Mean concentration (ng/mL) | 184.60 | 155.72 | 171.41 | 171.36 | 164.36 |
| Accuracy % | 111.57 | 94.11 | 103.60 | 103.57 | 99.34 |
| % CV | 6.36 | 4.03 | 2.97 | 6.59 | 7.16 |
| n | 3 | 3 | 3 | 3 | 3 |
| HighQC (1820 ng/mL) | |||||
| Species | Mouse | Rat | Dog | Monkey | Human |
| Mean concentration (ng/mL) | 1651.07 | 1792.99 | 1796.06 | 1667.92 | 1873.39 |
| Accuracy % | 90.72 | 98.52 | 98.69 | 91.64 | 102.93 |
| %CV | 7.51 | 6.08 | 3.52 | 6.14 | 2 |
| n | 3 | 3 | 3 | 3 | 3 |
| MMAF | QC |
|---|---|
| Mean concentration of the post-extraction spiked QC (ng/mL) | 198.84 |
| Mean concentration of the extracted QC samples (ng/mL) | 163.94 |
| Extraction recovery (%) | 82.45 |
| % CV | 2.12 |
| n | 3 |
| MMAF | Dilution QC (9100 ng/mL) |
| Mean concentration (ng/mL) | 8091.72 |
| Accuracy % | 88.92 |
| % CV | 9.02 |
| n | 3 |
| (a) Short-term stability (RT, 4 h) | Low QC (165.46 ng/mL) | High QC (1820 ng/mL) | ||||
| Incubation time (hr) | 0 (Control) | 4 | 0 (Control) | 4 | ||
| Mean Concentration (ng/mL) | 169.87 | 149.90 | 1970.02 | 1884.23 | ||
| Accuracy % | 102.67 | 90.60 | 108.24 | 103.53 | ||
| % CV | 2.08 | 3.05 | 5.20 | 3.27 | ||
| n | 3 | 3 | 3 | 3 | ||
| (b) Long-term stability (−80 °C, 4 weeks) | Low QC (165.46 ng/mL) | High QC (1820 ng/mL) | ||||
| Mean concentration (ng/mL) | 157.95 | 1657.96 | ||||
| Accuracy % | 95.46 | 91.10 | ||||
| % CV | 8.32 | 11.93 | ||||
| n | 3 | 3 | ||||
| (c) Freeze-thaw stability (−80 °C, three cycles) | Low QC (165.46 ng/mL) | High QC (1820 ng/mL) | ||||
| Mean concentration (ng/mL) | 157.95 | 1657.96 | ||||
| Accuracy % | 95.46 | 91.10 | ||||
| % CV | 8.32 | 11.93 | ||||
| n | 3 | 3 | ||||
| (d) Post-preparative stability (10 °C, 12 h) | Area ratio of 1st injection | Area ratio of 10th injection (after 12hr) | Change (%) | % CV | ||
| Low QC | 0.62 | 0.61 | 97.93 | 2.67 | ||
| High QC | 7.43 | 7.43 | 99.98 | 0.43 | ||
| Subject | Dose (mg/kg) | Cmax (ng/mL) | AUClast (min*ng/mL) | Clearance (CL) (mL/min/kg) | Vss (mL/kg) | Bioavailability (%) |
|---|---|---|---|---|---|---|
| MMAF IV (5 mg/kg) | 5 | 8276.76 | 65661.30 | 77.33 | 1057.13 | |
| MMAF PO (10 mg/kg) | 10 | N/D | N/D | 0 |
| Symbol | Metabolite | m/z | Retention Time (min) | Rat Liver Microsome | Human Liver Microsome | Pooled Rat Plasma (IV) | Pooled Rat Plasma (PO) |
|---|---|---|---|---|---|---|---|
| M1 | Oxidation-1 | 748.4624 | 21.50 | 0 | N/D | N/D | |
| M2 | Demethylation-1 | 718.4519 | 22.40 | 0 | 0 | N/D | N/D |
| M3 | Di-demethylation | 704.4362 | 22.96 | 0 | N/D | N/D | |
| M4 | Loss of C6H11NO | 619.3813 | 23.17 | 0 | 0 | N/D | N/D |
| M5 | Oxidation-2 | 748.4624 | 24.01 | 0 | N/D | N/D | |
| M6 | Demethylation-2 | 718.4519 | 24.55 | 0 | 0 | N/D | N/D |
| Parent | Parent | 732.4911 | 25.10 | 0 | 0 | 0 | 0 |
| M7 | Oxidation-3 | 748.4624 | 32.16 | 0 | 0 | N/D | N/D |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.-H.; Lee, B.i.; Byeon, J.-J.; Shin, S.-H.; Choi, J.; Park, Y.; Shin, Y.G. Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules 2019, 24, 2754. https://doi.org/10.3390/molecules24152754
Park M-H, Lee Bi, Byeon J-J, Shin S-H, Choi J, Park Y, Shin YG. Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules. 2019; 24(15):2754. https://doi.org/10.3390/molecules24152754
Chicago/Turabian StylePark, Min-Ho, Byeong ill Lee, Jin-Ju Byeon, Seok-Ho Shin, Jangmi Choi, Yuri Park, and Young G. Shin. 2019. "Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry" Molecules 24, no. 15: 2754. https://doi.org/10.3390/molecules24152754
APA StylePark, M. -H., Lee, B. i., Byeon, J. -J., Shin, S. -H., Choi, J., Park, Y., & Shin, Y. G. (2019). Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules, 24(15), 2754. https://doi.org/10.3390/molecules24152754