
Mammalian DNA Polymerase Kappa Activity and Specificity


Abstract
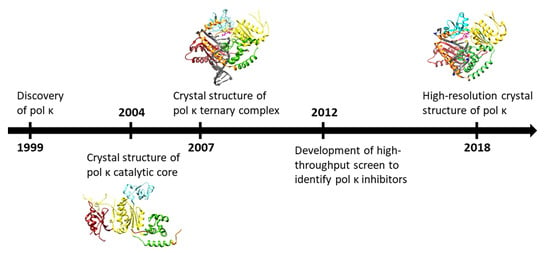
:
1. Introduction
2. Structure
3. Regulation
4. Fidelity and Selectivity
4.1. Role in TLS Extension Step
4.2. Pol Kappa Specificity and Cellular Functions
5. Roles of Pol κ Beyond Translesion Synthesis
6. Cancer-Associated Single-Nucleotide Polymorphisms (SNPs) in Human Pol κ
7. Inhibitors
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell. Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal Structure of a Y-Family DNA Polymerase in Action: A Mechanism for Error-Prone and Lesion-Bypass Replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed]
- Gruz, P.; Pisani, F.M.; Shimizu, M.; Yamada, M.; Hayashi, I.; Morikawa, K.; Nohmi, T. Synthetic activity of Sso DNA polymerase Y1, an archaeal DinB-like DNA polymerase, is stimulated by processivity factors proliferating cell nuclear antigen and replication factor C. J. Biol. Chem. 2001, 276, 47394–47401. [Google Scholar] [CrossRef] [PubMed]
- Niimi, N.; Sassa, A.; Katafuchi, A.; Gruz, P.; Fujimoto, H.; Bonala, R.R.; Johnson, F.; Ohta, T.; Nohmi, T. The steric gate amino acid tyrosine 112 is required for efficient mismatched-primer extension by human DNA polymerase kappa. Biochemistry 2009, 48, 4239–4246. [Google Scholar] [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Wagner, J.; Gruz, P.; Kim, S.R.; Yamada, M.; Matsui, K.; Fuchs, R.P.; Nohmi, T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell 1999, 4, 281–286. [Google Scholar] [CrossRef]
- Chandani, S.; Jacobs, C.; Loechler, E.L. Architecture of Y-family DNA polymerases relevant to translesion DNA synthesis as revealed in structural and molecular modeling studies. J. Nucleic Acids 2010, 2010, 784081. [Google Scholar] [CrossRef]
- Choi, J.Y.; Angel, K.C.; Guengerich, F.P. Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. J. Biol. Chem. 2006, 281, 21062–21072. [Google Scholar] [CrossRef]
- Huang, X.; Kolbanovskiy, A.; Wu, X.; Zhang, Y.; Wang, Z.; Zhuang, P.; Amin, S.; Geacintov, N.E. Effects of base sequence context on translesion synthesis past a bulky (+)-trans-anti-B[a]P-N2-dG lesion catalyzed by the Y-family polymerase pol kappa. Biochemistry 2003, 42, 2456–2466. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Godoy, V.G.; DeLaney, J.C.; Essigmann, J.M.; Walker, G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 2006, 439, 225–228. [Google Scholar] [CrossRef]
- Ogi, T.; Shinkai, Y.; Tanaka, K.; Ohmori, H. Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. PNAS 2002, 99, 15548–15553. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Zhang, Y.; Guo, D.; Wang, Z.; Amin, S.; Krzeminsky, J.; Louneva, N.; Geacintov, N.E. trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 2002, 277, 30488–30494. [Google Scholar] [CrossRef]
- Shen, X.; Sayer, J.M.; Kroth, H.; Ponten, I.; O’Donnell, M.; Woodgate, R.; Jerina, D.M.; Goodman, M.F. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J. Biol. Chem. 2002, 277, 5265–5274. [Google Scholar] [CrossRef]
- Suzuki, N.; Ohashi, E.; Kolbanovskiy, A.; Geacintov, N.E.; Grollman, A.P.; Ohmori, H.; Shibutani, S. Translesion synthesis by human DNA polymerase kappa on a DNA template containing a single stereoisomer of dG-(+)- or dG-(-)-anti-N(2)-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry 2002, 41, 6100–6106. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic. Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [Green Version]
- Boudsocq, F.; Kokoska, R.J.; Plosky, B.S.; Vaisman, A.; Ling, H.; Kunkel, T.A.; Yang, W.; Woodgate, R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J. Biol. Chem. 2004, 279, 32932–32940. [Google Scholar] [CrossRef]
- Pata, J.D. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta 2010, 1804, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.C.; Jackson, M.A.; Pata, J.D. Y-family polymerase conformation is a major determinant of fidelity and translesion specificity. Structure 2013, 21, 20–31. [Google Scholar] [CrossRef]
- Uljon, S.N.; Johnson, R.E.; Edwards, T.A.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Crystal Structure of the Catalytic Core of Human DNA Polymerase Kappa. Structure 2004, 12, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Lone, S.; Townson, S.A.; Uljon, S.N.; Johnson, R.E.; Brahma, A.; Nair, D.T.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Human DNA polymerase kappa encircles DNA: Implications for mismatch extension and lesion bypass. Mol. Cell 2007, 25, 601–614. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. 2.0 A resolution crystal structure of human polkappa reveals a new catalytic function of N-clasp in DNA replication. Sci Rep. 2018, 8, 15125. [Google Scholar] [CrossRef]
- Joyce, C.M.; Benkovic, S.J. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry 2004, 43, 14317–14324. [Google Scholar] [CrossRef]
- Berezhna, S.Y.; Gill, J.P.; Lamichhane, R.; Millar, D.P. Single-molecule Forster resonance energy transfer reveals an innate fidelity checkpoint in DNA polymerase I. J. Am. Chem. Soc. 2012, 134, 11261–11268. [Google Scholar] [CrossRef]
- Hohlbein, J.; Aigrain, L.; Craggs, T.D.; Bermek, O.; Potapova, O.; Shoolizadeh, P.; Grindley, N.D.; Joyce, C.M.; Kapanidis, A.N. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nat. Commun. 2013, 4, 2131. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.J.; Allen, W.J.; Sisamakis, E.; Kalinin, S.; Felekyan, S.; Widengren, J.; Waksman, G.; Seidel, C.A. dNTP-dependent conformational transitions in the fingers subdomain of Klentaq1 DNA polymerase: Insights into the role of the “nucleotide-binding” state. J. Biol. Chem. 2013, 288, 13575–13591. [Google Scholar] [CrossRef]
- Santoso, Y.; Joyce, C.M.; Potapova, O.; Le Reste, L.; Hohlbein, J.; Torella, J.P.; Grindley, N.D.; Kapanidis, A.N. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. PNAS 2010, 107, 715–720. [Google Scholar] [CrossRef]
- Velasco-Miguel, S.; Richardson, J.A.; Gerlach, V.L.; Lai, W.C.; Gao, T.; Russell, L.D.; Hladik, C.L.; White, C.L.; Friedberg, E.C. Constitutive and regulated expression of the mouse Dinb (Polkappa) gene encoding DNA polymerase kappa. DNA Repair (Amst) 2003, 2, 91–106. [Google Scholar] [CrossRef]
- Lemee, F.; Bavoux, C.; Pillaire, M.J.; Bieth, A.; Machado, C.R.; Pena, S.D.; Guimbaud, R.; Selves, J.; Hoffmann, J.S.; Cazaux, C. Characterization of promoter regulatory elements involved in downexpression of the DNA polymerase kappa in colorectal cancer. Oncogene 2007, 26, 3387–3394. [Google Scholar] [CrossRef]
- Zhu, H.; Fan, Y.; Shen, J.; Qi, H.; Shao, J. Characterization of human DNA polymerase kappa promoter in response to benzo[a]pyrene diol epoxide. Environ. Toxicol. Pharmacol. 2012, 33, 205–211. [Google Scholar] [CrossRef]
- Brauze, D.; Rawluszko, A.A. The effect of aryl hydrocarbon receptor ligands on the expression of polymerase (DNA directed) kappa (Polkappa), polymerase RNA II (DNA directed) polypeptide A (PolR2a), CYP1B1 and CYP1A1 genes in rat liver. Environ. Toxicol. Pharmacol. 2012, 34, 819–825. [Google Scholar] [CrossRef]
- Ogi, T.; Mimura, J.; Hikida, M.; Fujimoto, H.; Fujii-Kuriyama, Y.; Ohmori, H. Expression of human and mouse genes encoding polkappa: Testis-specific developmental regulation and AhR-dependent inducible transcription. Genes Cells 2001, 6, 943–953. [Google Scholar] [CrossRef]
- Bavoux, C.; Leopoldino, A.M.; Bergoglio, V.; Jiyang, O.W.; Ogi, T.; Bieth, A.; Judde, J.G.; Pena, S.D.; Poupon, M.F.; Helleday, T.; et al. Up-regulation of the error-prone DNA polymerase {kappa} promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005, 65, 325–330. [Google Scholar]
- Wang, J.; Kawamura, K.; Tada, Y.; Ohmori, H.; Kimura, H.; Sakiyama, S.; Tagawa, M. DNA Polymerase k, Implicated in Spontaneous and DNA Damage-induced Mutagenesis, Is Overexpressed in Lung Cancer. Cancer Res. 2001, 61, 5366–5369. [Google Scholar]
- Wang, H.; Wu, W.; Wang, H.W.; Wang, S.; Chen, Y.; Zhang, X.; Yang, J.; Zhao, S.; Ding, H.F.; Lu, D. Analysis of specialized DNA polymerases expression in human gliomas: Association with prognostic significance. Neuro Oncol. 2010, 12, 679–686. [Google Scholar] [CrossRef]
- Wang, Y.; Seimiya, M.; Kawamura, K.; Yu, L.; Ogi, T.; Takenaga, K.; Shishikura, T.; Nakagawara, A.; Sakiyama, S.; Tagawa, M.; et al. Elevated expression of DNA polymerase kappa in human lung cancer is associated with p53 inactivation: Negative regulation of POLK promoter activity by p53. Int. J. Oncol. 2004, 25, 161–165. [Google Scholar]
- Xie, W.; Yang, X.; Xu, M.; Jiang, T. Structural insights into the assembly of human translesion polymerase complexes. Protein Cell 2012, 3, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.J.; Colnaghi, L.; Huang, T.T. Dysregulation of DNA polymerase kappa recruitment to replication forks results in genomic instability. EMBO J. 2012, 31, 908–918. [Google Scholar] [CrossRef]
- Guo, C.; Tang, T.S.; Bienko, M.; Dikic, I.; Friedberg, E.C. Requirements for the interaction of mouse Polkappa with ubiquitin and its biological significance. J. Biol. Chem. 2008, 283, 4658–4664. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, S.; Prakash, L. The human DINB1 gene encodes the DNA polymerase Pol theta. PNAS 2000, 97, 3838–3843. [Google Scholar] [CrossRef]
- Ohashi, E.; Bebenek, K.; Matsuda, T.; Feaver, W.J.; Gerlach, V.L.; Friedberg, E.C.; Ohmori, H.; Kunkel, T.A. Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J. Biol. Chem. 2000, 275, 39678–39684. [Google Scholar] [CrossRef]
- Nevin, P.; Lu, X.; Zhang, K.; Engen, J.R.; Beuning, P.J. Noncognate DNA damage prevents the formation of the active conformation of the Y-family DNA polymerases DinB and DNA polymerase kappa. FEBS J. 2015, 282, 2646–2660. [Google Scholar] [CrossRef]
- Zhao, L.; Pence, M.G.; Eoff, R.L.; Yuan, S.; Fercu, C.A.; Guengerich, F.P. Elucidation of kinetic mechanisms of human translesion DNA polymerase kappa using tryptophan mutants. FEBS J. 2014, 281, 4394–4410. [Google Scholar] [CrossRef]
- Fischhaber, P.L.; Gerlach, V.L.; Feaver, W.J.; Hatahet, Z.; Wallace, S.S.; Friedberg, E.C. Human DNA polymerase kappa bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J. Biol. Chem. 2002, 277, 37604–37611. [Google Scholar] [CrossRef]
- Levine, R.L.; Miller, H.; Grollman, A.; Ohashi, E.; Ohmori, H.; Masutani, C.; Hanaoka, F.; Moriya, M. Translesion DNA synthesis catalyzed by human pol eta and pol kappa across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 2001, 276, 18717–18721. [Google Scholar] [CrossRef]
- Wolfle, W.T.; Washington, M.T.; Prakash, L.; Prakash, S. Human DNA polymerase kappa uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003, 17, 2191–2199. [Google Scholar] [CrossRef]
- Walsh, J.M.; Ippoliti, P.J.; Ronayne, E.A.; Rozners, E.; Beuning, P.J. Discrimination against major groove adducts by Y-family polymerases of the DinB subfamily. DNA Repair (Amst) 2013, 12, 713–722. [Google Scholar] [CrossRef]
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Du, H.; Wang, P.; Li, L.; Wang, Y. Repair and translesion synthesis of O (6)-alkylguanine DNA lesions in human cells. J. Biol. Chem. 2019, 294, 11144–11153. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Guo, D.; Rechkoblit, O.; Wang, Z. Activities of human DNA polymerase kappa in response to the major benzo[a]pyrene DNA adduct: Error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair (Amst) 2002, 1, 559–569. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Guo, C. Effects of the N terminus of mouse DNA polymerase kappa on the bypass of a guanine-benzo[a]pyrenyl adduct. J. Biochem. 2016, 159, 471–479. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Tang, T.S.; Zhang, H.; Wang, Z.; Friedberg, E.; Yang, W.; Guo, C. Variants of mouse DNA polymerase kappa reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. PNAS 2014, 111, 1789–1794. [Google Scholar] [CrossRef]
- Sassa, A.; Niimi, N.; Fujimoto, H.; Katafuchi, A.; Gruz, P.; Yasui, M.; Gupta, R.C.; Johnson, F.; Ohta, T.; Nohmi, T. Phenylalanine 171 is a molecular brake for translesion synthesis across benzo[a]pyrene-guanine adducts by human DNA polymerase kappa. Mutat. Res. 2011, 718, 10–17. [Google Scholar] [CrossRef]
- Zhao, L.; Washington, M.T. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes (Basel) 2017, 8, 24. [Google Scholar] [CrossRef]
- Yuan, B.; Cao, H.; Jiang, Y.; Hong, H.; Wang, Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2’-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. PNAS 2008, 105, 8679–8684. [Google Scholar] [CrossRef]
- Wu, J.; Wang, P.; Li, L.; Williams, N.L.; Ji, D.; Zahurancik, W.J.; You, C.; Wang, J.; Suo, Z.; Wang, Y. Replication studies of carboxymethylated DNA lesions in human cells. Nucleic Acids Res. 2017, 45, 7276–7284. [Google Scholar] [CrossRef] [Green Version]
- Gowda, A.S.; Lee, M.; Spratt, T.E. N2 -Substituted 2’-Deoxyguanosine Triphosphate Derivatives as Selective Substrates for Human DNA Polymerase kappa. Angew. Chem. Int. Ed. Engl. 2017, 56, 2628–2631. [Google Scholar] [CrossRef]
- Andersen, N.; Wang, J.; Wang, P.; Jiang, Y.; Wang, Y. In-vitro replication studies on O(2)-methylthymidine and O(4)-methylthymidine. Chem. Res. Toxicol. 2012, 25, 2523–2531. [Google Scholar] [CrossRef]
- Andersen, N.; Wang, P.; Wang, Y. Replication across regioisomeric ethylated thymidine lesions by purified DNA polymerases. Chem. Res. Toxicol. 2013, 26, 1730–1738. [Google Scholar] [CrossRef]
- Gowda, A.S.P.; Spratt, T.E. DNA Polymerases eta and zeta Combine to Bypass O-2-[4-(3-Pyridyl)-4-oxobutyl]thymine, a DNA Adduct Formed from Tobacco Carcinogens. Chem. Res. Toxicol. 2016, 29, 303–316. [Google Scholar] [CrossRef]
- Wilson, K.A.; Holland, C.D.; Wetmore, S.D. Uncovering a unique approach for damaged DNA replication: A computational investigation of a mutagenic tobacco-derived thymine lesion. Nucleic Acids Res. 2019, 47, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Zang, H.; Angel, K.C.; Kozekov, I.D.; Goodenough, A.K.; Rizzo, C.J.; Guengerich, F.P. Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 2006, 19, 879–886. [Google Scholar] [CrossRef]
- Wickramaratne, S.; Boldry, E.J.; Buehler, C.; Wang, Y.C.; Distefano, M.D.; Tretyakova, N.Y. Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine. J. Biol. Chem. 2015, 290, 775–787. [Google Scholar] [CrossRef]
- Basu, A.K.; Pande, P.; Bose, A. Translesion Synthesis of 2’-Deoxyguanosine Lesions by Eukaryotic DNA Polymerases. Chem. Res. Toxicol. 2017, 30, 61–72. [Google Scholar] [CrossRef]
- Roy, U.; Scharer, O.D. Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair (Amst) 2016, 44, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, A.V.; Bondarenko, K.A.; Makarova, A.V. Non-bulky Lesions in Human DNA: The Ways of Formation, Repair, and Replication. Acta Nat. 2017, 9, 12–26. [Google Scholar] [CrossRef]
- Yagi, T.; Fujikawa, Y.; Sawai, T.; Takamura-Enya, T.; Ito-Harashima, S.; Kawanishi, M. Error-Prone and Error-Free Translesion DNA Synthesis over Site-Specifically Created DNA Adducts of Aryl Hydrocarbons (3-Nitrobenzanthrone and 4-Aminobiphenyl). Toxicol. Res. 2017, 33, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Kino, K.; Hirao-Suzuki, M.; Morikawa, M.; Sakaga, A.; Miyazawa, H. Generation, repair and replication of guanine oxidation products. Genes Environ. 2017, 39, 21. [Google Scholar] [CrossRef]
- Yockey, O.P.; Jha, V.; Ghodke, P.P.; Xu, T.; Xu, W.; Ling, H.; Pradeepkumar, P.I.; Zhao, L. Mechanism of Error-Free DNA Replication Past Lucidin-Derived DNA Damage by Human DNA Polymerase kappa. Chem. Res. Toxicol. 2017, 30, 2023–2032. [Google Scholar] [CrossRef]
- Gerlach, V.L.; Feaver, W.J.; Fischhaber, P.L.; Friedberg, E.C. Purification and characterization of pol kappa, a DNA polymerase encoded by the human DINB1 gene. J. Biol. Chem. 2001, 276, 92–98. [Google Scholar] [CrossRef]
- Washington, M.T.; Johnson, R.E.; Prakash, L.; Prakash, S. Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. PNAS 2002, 99, 1910–1914. [Google Scholar] [CrossRef]
- Wolfle, W.T.; Washington, M.T.; Kool, E.T.; Spratt, T.E.; Helquist, S.A.; Prakash, L.; Prakash, S. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase kappa. Mol. Cell Biol. 2005, 25, 7137–7143. [Google Scholar] [CrossRef]
- Antczak, N.M.; Packer, M.R.; Lu, X.; Zhang, K.; Beuning, P.J. Human Y-Family DNA Polymerase kappa Is More Tolerant to Changes in Its Active Site Loop than Its Ortholog Escherichia coli DinB. Chem. Res. Toxicol. 2017. [Google Scholar] [CrossRef]
- Ketkar, A.; Maddukuri, L.; Penthala, N.R.; Reed, M.R.; Zafar, M.K.; Crooks, P.A.; Eoff, R.L. Inhibition of Human DNA Polymerases Eta and Kappa by Indole-Derived Molecules Occurs through Distinct Mechanisms. ACS Chem. Biol. 2019, 14, 1337–1351. [Google Scholar] [CrossRef]
- Nevin, P.; Engen, J.R.; Beuning, P.J. Steric gate residues of Y-family DNA polymerases DinB and pol kappa are crucial for dNTP-induced conformational change. DNA Repair (Amst) 2015, 29, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Ribonucleotide discrimination by translesion synthesis DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 382–402. [Google Scholar] [CrossRef]
- Haracska, L.; Prakash, L.; Prakash, S. Role of human DNA polymerase kappa as an extender in translesion synthesis. PNAS 2002, 99, 16000–16005. [Google Scholar] [CrossRef]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef]
- Carlson, K.D.; Johnson, R.E.; Prakash, L.; Prakash, S.; Washington, M.T. Human DNA polymerase kappa forms nonproductive complexes with matched primer termini but not with mismatched primer termini. PNAS 2006, 103, 15776–15781. [Google Scholar] [CrossRef]
- Dong, H.; Bonala, R.R.; Suzuki, N.; Johnson, F.; Grollman, A.P.; Shibutani, S. Mutagenic potential of benzo[a]pyrene-derived DNA adducts positioned in codon 273 of the human P53 gene. Biochemistry 2004, 43, 15922–15928. [Google Scholar] [CrossRef]
- Choi, J.Y.; Guengerich, F.P. Analysis of the Effect of Bulk at N2-Alkylguanine DNA Adducts on Catalytic Efficiency and Fidelity of the Processive DNA Polymerases Bacteriophage T7 Exonuclease- and HIV-1 Reverse Transcriptase. J. Biol. Chem. 2004, 279, 19217–19229. [Google Scholar] [CrossRef] [Green Version]
- Chiapperino, D.; Kroth, H.; Kramarczuk, I.H.; Sayer, J.M.; Masutani, C.; Hanaoka, F.; Jerina, D.M.; Cheh, A.M. Preferential misincorporation of purine nucleotides by human DNA polymerase eta opposite benzo[a]pyrene 7,8-diol 9,10-epoxide deoxyguanosine adducts. J. Biol. Chem. 2002, 277, 11765–11771. [Google Scholar] [CrossRef]
- Frank, E.G.; Sayer, J.M.; Kroth, H.; Ohashi, E.; Ohmori, H.; Jerina, D.M.; Woodgate, R. Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase iota. Nucleic Acids Res. 2002, 30, 5284–5292. [Google Scholar] [CrossRef] [Green Version]
- Klarer, A.C.; Stallons, L.J.; Burke, T.J.; Skaggs, R.L.; McGregor, W.G. DNA polymerase eta participates in the mutagenic bypass of adducts induced by benzo[a]pyrene diol epoxide in mammalian cells. PLoS ONE 2012, 7, e39596. [Google Scholar] [CrossRef]
- Xie, Z.; Braithwaite, E.; Guo, D.; Zhao, B.; Geacintov, N.E.; Wang, Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: Requirement for DNA polymerase zeta and involvement of DNA polymerase eta. Biochemistry 2003, 42, 11253–11262. [Google Scholar] [CrossRef]
- Jha, V.; Bian, C.; Xing, G.; Ling, H. Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase kappa. Nucleic Acids Res. 2016, 44, 4957–4967. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. Structural basis of accurate replication beyond a bulky major benzo[a]pyrene adduct by human DNA polymerase kappa. DNA Repair (Amst) 2017, 49, 43–50. [Google Scholar] [CrossRef]
- Sassa, A.; Suzuki, T.; Kanemaru, Y.; Niimi, N.; Fujimoto, H.; Katafuchi, A.; Gruz, P.; Yasui, M.; Gupta, R.C.; Johnson, F.; et al. In vivo evidence that phenylalanine 171 acts as a molecular brake for translesion DNA synthesis across benzo[a]pyrene DNA adducts by human DNA polymerase kappa. DNA Repair (Amst) 2014, 15, 21–28. [Google Scholar] [CrossRef]
- Kanemaru, Y.; Suzuki, T.; Niimi, N.; Gruz, P.; Matsumoto, K.; Adachi, N.; Honma, M.; Nohmi, T. Catalytic and non-catalytic roles of DNA polymerase kappa in the protection of human cells against genotoxic stresses. Environ. Mol. Mutagen. 2015, 56, 650–662. [Google Scholar] [CrossRef]
- Kanemaru, Y.; Suzuki, T.; Sassa, A.; Matsumoto, K.; Adachi, N.; Honma, M.; Numazawa, S.; Nohmi, T. DNA polymerase kappa protects human cells against MMC-induced genotoxicity through error-free translesion DNA synthesis. Genes Environ. 2017, 39, 6. [Google Scholar] [CrossRef]
- Avkin, S.; Goldsmith, M.; Velasco-Miguel, S.; Geacintov, N.; Friedberg, E.C.; Livneh, Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase kappa. J. Biol. Chem. 2004, 279, 53298–53305. [Google Scholar] [CrossRef]
- Masumura, K.; Toyoda-Hokaiwado, N.; Niimi, N.; Gruz, P.; Wada, N.A.; Takeiri, A.; Jishage, K.I.; Mishima, M.; Nohmi, T. Limited ability of DNA polymerase kappa to suppress benzo[a]pyrene-induced genotoxicity in vivo. Environ. Mol. Mutagen. 2017, 58, 644–653. [Google Scholar] [CrossRef]
- Hakura, A.; Sui, H.; Sonoda, J.; Matsuda, T.; Nohmi, T. DNA polymerase kappa counteracts inflammation-induced mutagenesis in multiple organs of mice. Environ. Mol. Mutagen. 2019, 60, 320–330. [Google Scholar] [CrossRef]
- Takeiri, A.; Wada, N.A.; Motoyama, S.; Matsuzaki, K.; Tateishi, H.; Matsumoto, K.; Niimi, N.; Sassa, A.; Gruz, P.; Masumura, K.; et al. In vivo evidence that DNA polymerase kappa is responsible for error-free bypass across DNA cross-links induced by mitomycin C. DNA Repair (Amst) 2014, 24, 113–121. [Google Scholar] [CrossRef]
- Bargonetti, J.; Champeil, E.; Tomasz, M. Differential toxicity of DNA adducts of mitomycin C. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Rycenga, H.B.; Long, D.T. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol. 2018, 41, 20–26. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Szikriszt, B.; Poti, A.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnar, J.; Ribli, D.; Szeltner, Z.; Tusnady, G.E.; Csabai, I.; et al. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. Structural Basis for Human DNA Polymerase Kappa to Bypass Cisplatin Intrastrand Cross-Link (Pt-GG) Lesion as an Efficient and Accurate Extender. J. Mol. Biol. 2018, 430, 1577–1589. [Google Scholar] [CrossRef]
- Ho, T.V.; Guainazzi, A.; Derkunt, S.B.; Enoiu, M.; Scharer, O.D. Structure-dependent bypass of DNA interstrand crosslinks by translesion synthesis polymerases. Nucleic Acids Res. 2011, 39, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Enoiu, M.; Jiricny, J.; Scharer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef]
- Williams, H.L.; Gottesman, M.E.; Gautier, J. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 2012, 47, 140–147. [Google Scholar] [CrossRef]
- Zhuo, M.; Gorgun, M.F.; Englander, E.W. Translesion Synthesis DNA Polymerase Kappa Is Indispensable for DNA Repair Synthesis in Cisplatin Exposed Dorsal Root Ganglion Neurons. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Minko, I.G.; Yamanaka, K.; Kozekov, I.D.; Kozekova, A.; Indiani, C.; O’Donnell, M.E.; Jiang, Q.; Goodman, M.F.; Rizzo, C.J.; Lloyd, R.S. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008, 21, 1983–1990. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef]
- Boysen, G.; Pachkowski, B.F.; Nakamura, J.; Swenberg, J.A. The formation and biological significance of N7-guanine adducts. Mutat. Res. 2009, 678, 76–94. [Google Scholar] [CrossRef] [Green Version]
- Klapacz, J.; Pottenger, L.H.; Engelward, B.P.; Heinen, C.D.; Johnson, G.E.; Clewell, R.A.; Carmichael, P.L.; Adeleye, Y.; Andersen, M.E. Contributions of DNA repair and damage response pathways to the non-linear genotoxic responses of alkylating agents. Mutat. Res. Rev. Mutat. Res. 2016, 767, 77–91. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Yi, C.; He, C. DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol. 2013, 5, a012575. [Google Scholar] [CrossRef]
- Gates, K.S. Structural biology: FaPy lesions and DNA mutations. Nat. Chem. Biol. 2013, 9, 412–414. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef]
- Banerjee, S.; Chakraborty, S.; Jacinto, M.P.; Paul, M.D.; Balster, M.V.; Greenberg, M.M. Probing Enhanced Double-Strand Break Formation at Abasic Sites within Clustered Lesions in Nucleosome Core Particles. Biochemistry 2017, 56, 14–21. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Yu, S.L.; Prakash, S.; Prakash, L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol. Cell Biol. 2007, 27, 7198–7205. [Google Scholar] [CrossRef]
- Takenaka, K.; Ogi, T.; Okada, T.; Sonoda, E.; Guo, C.; Friedberg, E.C.; Takeda, S. Involvement of vertebrate Polkappa in translesion DNA synthesis across DNA monoalkylation damage. J. Biol. Chem. 2006, 281, 2000–2004. [Google Scholar] [CrossRef]
- Takenaka, K.; Miki, Y. Introduction and characterization of a polymerase-dead point mutation into the POLK gene in vertebrates. FEBS Lett 2009, 583, 661–664. [Google Scholar] [CrossRef]
- Wit, N.; Buoninfante, O.A.; van den Berk, P.C.; Jansen, J.G.; Hogenbirk, M.A.; de Wind, N.; Jacobs, H. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015, 43, 282–294. [Google Scholar] [CrossRef]
- Plosky, B.S.; Frank, E.G.; Berry, D.A.; Vennall, G.P.; McDonald, J.P.; Woodgate, R. Eukaryotic Y-family polymerases bypass a 3-methyl-2’-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008, 36, 2152–2162. [Google Scholar] [CrossRef]
- Yoon, J.H.; Roy Choudhury, J.; Park, J.; Prakash, S.; Prakash, L. Translesion synthesis DNA polymerases promote error-free replication through the minor-groove DNA adduct 3-deaza-3-methyladenine. J. Biol. Chem. 2017, 292, 18682–18688. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, E.; Ogi, T.; Kusumoto, R.; Iwai, S.; Masutani, C.; Hanaoka, F.; Ohmori, H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000, 14, 1589–1594. [Google Scholar]
- Sherrer, S.M.; Fiala, K.A.; Fowler, J.D.; Newmister, S.A.; Pryor, J.M.; Suo, Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2011, 39, 609–622. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lim, S.; Kim, E.J.; Jo, A.; Guengerich, F.P. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J. Mol. Biol. 2010, 404, 34–44. [Google Scholar] [CrossRef]
- Hile, S.E.; Eckert, K.A. DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 2008, 36, 688–696. [Google Scholar] [CrossRef]
- Hile, S.E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Beyond translesion synthesis: Polymerase kappa fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012, 40, 1636–1647. [Google Scholar] [CrossRef]
- Barnes, R.P.; Hile, S.E.; Lee, M.Y.; Eckert, K.A. DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst) 2017, 57, 1–11. [Google Scholar] [CrossRef]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef]
- Eddy, S.; Tillman, M.; Maddukuri, L.; Ketkar, A.; Zafar, M.K.; Eoff, R.L. Human Translesion Polymerase kappa Exhibits Enhanced Activity and Reduced Fidelity Two Nucleotides from G-Quadruplex DNA. Biochemistry 2016, 55, 5218–5229. [Google Scholar] [CrossRef]
- Ogi, T.; Lehmann, A.R. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006, 8, 640–642. [Google Scholar] [CrossRef]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef]
- Bergoglio, V.; Bavoux, C.; Verbiest, V.; Hoffmann, J.S.; Cazaux, C. Localisation of human DNA polymerase kappa to replication foci. J. Cell Sci. 2002, 115, 4413–4418. [Google Scholar] [CrossRef]
- Bi, X.; Barkley, L.R.; Slater, D.M.; Tateishi, S.; Yamaizumi, M.; Ohmori, H.; Vaziri, C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell Biol. 2006, 26, 3527–3540. [Google Scholar] [CrossRef]
- Betous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef]
- Tonzi, P.; Yin, Y.; Lee, C.W.T.; Rothenberg, E.; Huang, T.T. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife 2018, 7. [Google Scholar] [CrossRef]
- Yadav, S.; Mukhopadhyay, S.; Anbalagan, M.; Makridakis, N. Somatic Mutations in Catalytic Core of POLK Reported in Prostate Cancer Alter Translesion DNA Synthesis. Hum. Mutat. 2015, 36, 873–880. [Google Scholar] [CrossRef]
- Dai, Z.J.; Liu, X.H.; Ma, Y.F.; Kang, H.F.; Jin, T.B.; Dai, Z.M.; Guan, H.T.; Wang, M.; Liu, K.; Dai, C.; et al. Association Between Single Nucleotide Polymorphisms in DNA Polymerase Kappa Gene and Breast Cancer Risk in Chinese Han Population: A STROBE-Compliant Observational Study. Medicine (Baltimore) 2016, 95, e2466. [Google Scholar] [CrossRef]
- Shao, M.; Jin, B.; Niu, Y.; Ye, J.; Lu, D.; Han, B. Association of POLK polymorphisms with platinum-based chemotherapy response and severe toxicity in non-small cell lung cancer patients. Cell Biochem. Biophys. 2014, 70, 1227–1237. [Google Scholar] [CrossRef]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair (Amst) 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Song, I.; Kim, E.J.; Kim, I.H.; Park, E.M.; Lee, K.E.; Shin, J.H.; Guengerich, F.P.; Choi, J.Y. Biochemical characterization of eight genetic variants of human DNA polymerase kappa involved in error-free bypass across bulky N(2)-guanyl DNA adducts. Chem. Res. Toxicol. 2014, 27, 919–930. [Google Scholar] [CrossRef]
- Antczak, N.M.; Walker, A.R.; Stern, H.R.; Leddin, E.M.; Palad, C.; Coulther, T.A.; Swett, R.J.; Cisneros, G.A.; Beuning, P.J. Characterization of Nine Cancer-Associated Variants in Human DNA Polymerase kappa. Chem. Res. Toxicol. 2018, 31, 697–711. [Google Scholar] [CrossRef]
- Kim, J.K.; Yeom, M.; Hong, J.K.; Song, I.; Lee, Y.S.; Guengerich, F.P.; Choi, J.Y. Six Germline Genetic Variations Impair the Translesion Synthesis Activity of Human DNA Polymerase kappa. Chem. Res. Toxicol. 2016, 29, 1741–1754. [Google Scholar] [CrossRef]
- Han, J.; Haiman, C.; Niu, T.; Guo, Q.; Cox, D.G.; Willett, W.C.; Hankinson, S.E.; Hunter, D.J. Genetic variation in DNA repair pathway genes and premenopausal breast cancer risk. Breast Cancer Res. Treat. 2009, 115, 613–622. [Google Scholar] [CrossRef]
- Dorjsuren, D.; Wilson, D.M., 3rd; Beard, W.A.; McDonald, J.P.; Austin, C.P.; Woodgate, R.; Wilson, S.H.; Simeonov, A. A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 2009, 37, e128. [Google Scholar] [CrossRef]
- Yamanaka, K.; Dorjsuren, D.; Eoff, R.L.; Egli, M.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Lloyd, R.S. A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase kappa. PLoS ONE 2012, 7, e45032. [Google Scholar] [CrossRef]
- Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Yamanaka, K.; Banerjee, S.; Egli, M.; Choi, J.Y.; Lloyd, R.S.; Eoff, R.L. Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity. Chem. Res. Toxicol. 2013, 26, 221–232. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | ID | Effects | Domain | Tumor Site | References |
|---|---|---|---|---|---|
| L21F | rs3104729 | 30-fold decrease in incorporation opposite N2-CH2-(9-anthracenyl)-dG (N2-CH2-Anth-dG) | N-clasp | Prostate | [147,148] |
| E29K | Decreased insertion opposite abasic site (2–20×) | N-clasp | Prostate, early onset | [143] | |
| I39T | rs3094258 | Similar activity to WT with several types of DNA damage | N-clasp | Prostate, Melanoma | [147,148] |
| T44M | Lesion-specific reduction in activity; reduced activity with N2-CH2-Anth-dG, O6-Me-dG and abasic sites | N-clasp | [149] | ||
| S137S | Synonymous | Fingers | Prostate | [143] | |
| G154E | COSM3856305 | Decreased activity opposite model abasic site; pathogenic | Fingers | Prostate early onset, stomach | [143] |
| F155S | Decreased activity on model abasic site | Fingers | Prostate | [143] | |
| P169T | rs148385845 | Slight decrease in activity on undamaged DNA | Fingers | Lung a | [148] |
| F171F | Synonymous | Palm | Prostate | [143] | |
| D189G | rs111689950 | Impaired for extension step of TLS | Palm | [147] | |
| F192C | rs150515841 | Slight increase in activity with N2-furfuryl-dG-containing templates | Palm | [148] | |
| T205I | Palm | Prostate, early onset | [143] | ||
| R219I | rs3104717 | Slight decrease in activity | Palm | Prostate | [147,148] |
| R219X | rs3094265 b | Inactive | Palm | [147] | |
| R246X | COSM3073601 | 5-10-fold less active with 8-oxo-dG-, N2-CH2-Anth-dG-, O6-Me-dG- and abasic-containing templates | Stomach | [149] | |
| E292K | rs142203892 | Similar activity as WT | Palm | [148] | |
| R298H | rs151251843 | Less active than WT on several different lesions | Palm | Large intestine a | [148,149] |
| A329A | rs3213801 | Synonymous, not meaningfully associated with breast cancer risk; more likely to respond to Pt-based chemotherapy | Palm | Breast | [144,145] |
| E419G | rs111584802 | 20-fold decrease in kcat/Km on dG and 670-fold decrease on N2-CH2-Anth-dG, extension defect | Little finger c | [147] | |
| E419E | Synonymous | Little finger | Prostate | [143] | |
| S423R | rs35257416 COSM6752124 | 1.6-fold more efficient than WT, 2-fold increased DNA binding affinity, pathogenic | Little finger | Melanoma, large intestine | [147,148] |
| A428A | COSM1070129 | Synonymous | Little finger | Endometrium, Prostate | [143] |
| E430K | Little finger | Prostate | [143] | ||
| E430G | Low activity on AP site | Little finger | Prostate | [143] | |
| Y432S | rs77612491 | Less active on undamaged and damaged DNA, extension defect, decreased DNA binding affinity | Little finger | Melanoma | [147,148] |
| L442F | Low activity on AP site | Little finger | Prostate, early onset | [143] | |
| Q447Q | Synonymous | Little finger | Prostate | [143] | |
| E449K | Low activity on AP site, low fidelity | Little finger | Prostate | [143] | |
| K461E | Little finger | Prostate | [143] | ||
| A471V | rs149894654 | Moderate decrease in activity | Little finger | [149] | |
| T473A | rs186798689 | Decreased activity on undamaged and damaged DNA | Little finger | [149] | |
| I487T | Little finger | Prostate | [143] | ||
| R512W | Decreased activity on undamaged and damaged DNA | Little finger | [149] | ||
| S528N | Prostate | [143] | |||
| D551N | Prostate | [143] | |||
| K564K | Synonymous | Prostate | [143] | ||
| D581N | Prostate | [143] | |||
| S678F | Prostate | [143] | |||
| L731F | Prostate | [143] | |||
| P861P | Prostate | [143] | |||
| D866E | Prostate | [143] | |||
| intron | rs10077427 | Contributes to breast cancer risk, more likely to have progesterone receptor-positive tumors; Decreased progression-free survival with Pt-based chemotherapy | Breast | [144,145] | |
| intron | rs5744533 | Contributes to breast cancer risk, protective in postmenopausal women, no correlation with clinical phenotypes; More likely to respond to Pt-based chemotherapy | Breast | [144,145] | |
| intron | rs3756558 | Breast | [150] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stern, H.R.; Sefcikova, J.; Chaparro, V.E.; Beuning, P.J. Mammalian DNA Polymerase Kappa Activity and Specificity. Molecules 2019, 24, 2805. https://doi.org/10.3390/molecules24152805
Stern HR, Sefcikova J, Chaparro VE, Beuning PJ. Mammalian DNA Polymerase Kappa Activity and Specificity. Molecules. 2019; 24(15):2805. https://doi.org/10.3390/molecules24152805
Chicago/Turabian StyleStern, Hannah R., Jana Sefcikova, Victoria E. Chaparro, and Penny J. Beuning. 2019. "Mammalian DNA Polymerase Kappa Activity and Specificity" Molecules 24, no. 15: 2805. https://doi.org/10.3390/molecules24152805
APA StyleStern, H. R., Sefcikova, J., Chaparro, V. E., & Beuning, P. J. (2019). Mammalian DNA Polymerase Kappa Activity and Specificity. Molecules, 24(15), 2805. https://doi.org/10.3390/molecules24152805