
Synthesis and Antiproliferative Activity of Novel A-Ring Cleaved Glycyrrhetinic Acid Derivatives




Abstract
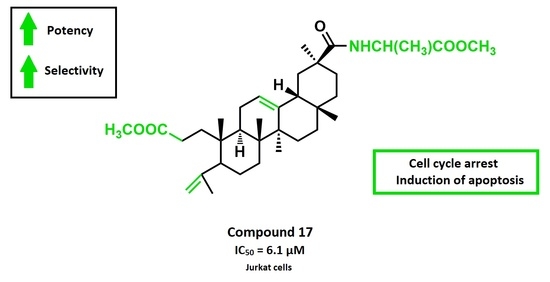
:
1. Introduction
2. Results and Discussion
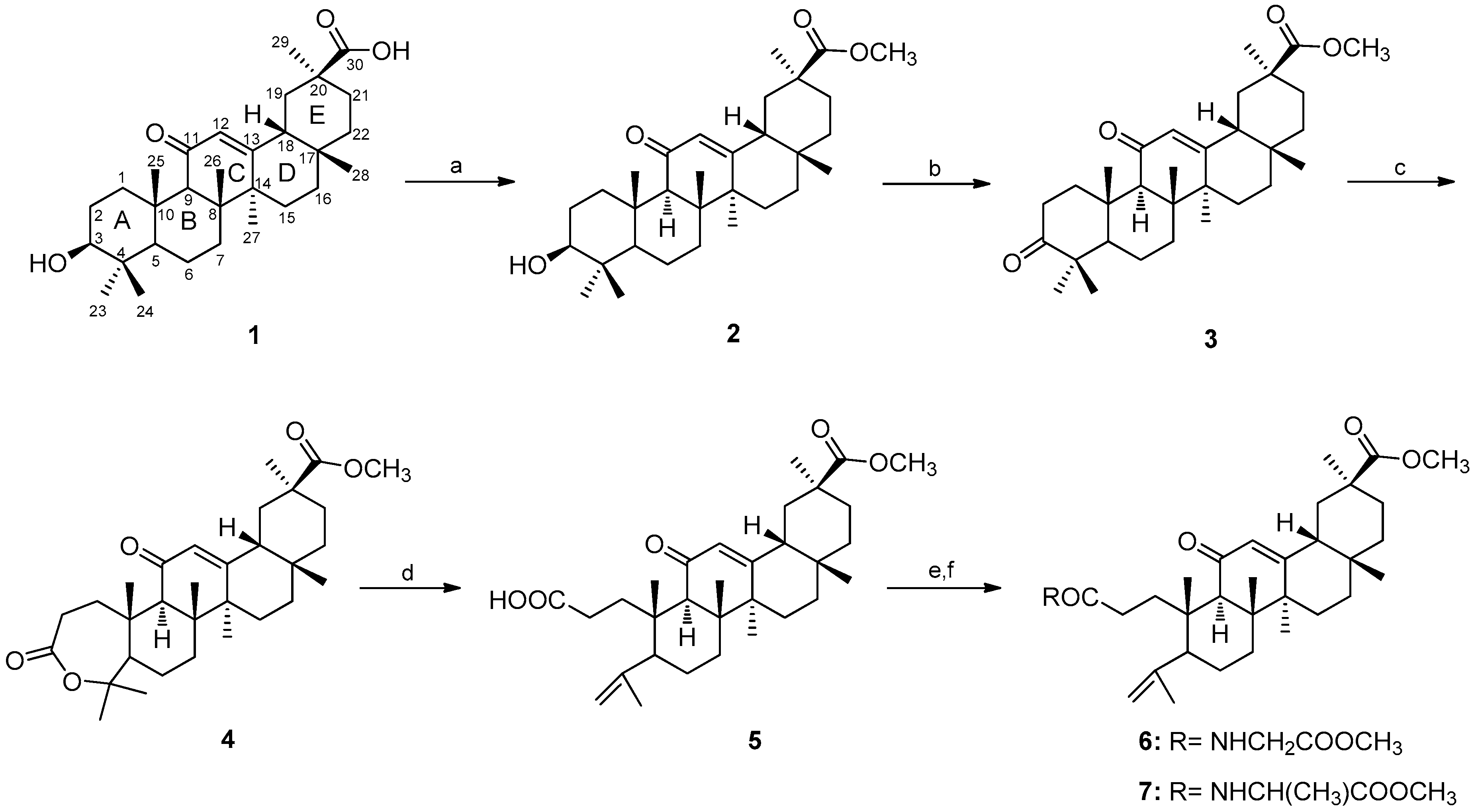
2.1. Chemistry
2.2. Biology
2.2.1. Antiproliferative Activity
2.2.2. Analysis of Cell Cycle Distribution and Apoptosis
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. Cell Culture
3.2.2. Antiproliferative Activity Assays
3.2.3. Cell Cycle Analysis
3.2.4. Annexin V-FITC/PI Flow Cytometry Assay
3.2.5. Hoechst 33342 Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Moreira, V.M.; Goncalves, B.M.F.; Leal, A.S.; Jing, Y.K. Ursane-type pentacyclic triterpenoids as useful platforms to discover anticancer drugs. Nat. Prod. Rep. 2012, 29, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Leal, A.S.; Alho, D.P.S.; Goncalves, B.M.F.; Valdeira, A.S.; Mendes, V.I.S.; Jing, Y.K. Highlights of pentacyclic triterpenoids in the cancer settings. In Studies in Natural Products Chemistry; AttaUrRahman, F.R.S., Ed.; Elsevier Science Bv: Amsterdam, The Netherlands, 2014; Volume 41, pp. 33–73. [Google Scholar]
- Chudzik, M.; Korzonek-Szlacheta, I.; Krol, W. Triterpenes as potentially cytotoxic compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Leal, A.S.; Valdeira, A.S.; Goncalves, B.M.F.; Alho, D.P.S.; Figueiredo, S.A.C.; Silvestre, S.M.; Mendes, V.I.S. Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur. J. Med. Chem. 2017, 142, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Schwarz, S.; Per, V.; Kowitsch, A.; Siewert, B.; Csuk, R. Incorporation of a michael acceptor enhances the antitumor activity of triterpenoic acids. Eur. J. Med. Chem. 2015, 101, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, J.; Heller, L.; Csuk, R. Targeting cancer cells with oleanolic and ursolic acid derived hydroxamates. Bio Org. Med. Chem. Lett. 2016, 26, 907–909. [Google Scholar] [CrossRef]
- Goncalves, B.M.F.; Salvador, J.A.R.; Marin, S.; Cascante, M. Synthesis and anticancer activity of novel fluorinated asiatic acid derivatives. Eur. J. Med. Chem. 2016, 114, 101–117. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Urea derivates of ursolic, oleanolic and maslinic acid induce apoptosis and are selective cytotoxic for several human tumor cell lines. Eur. J. Med. Chem. 2016, 119, 1–16. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine b conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.; Khalitova, R.; Nedopekina, D.; Dzhemileva, L.; Yunusbaeva, M.; Odinokov, V.; D’Yakonov, V.; Dzhemilev, U. Synthesis and evaluation of anticancer activities of novel c-28 guanidine-functionalized triterpene acid derivatives. Molecules 2018, 23, 22. [Google Scholar] [CrossRef] [PubMed]
- Valdeira, A.S.C.; Ritt, D.A.; Morrison, D.K.; McMahon, J.B.; Gustafson, K.R.; Salvador, J.A.R. Synthesis and biological evaluation of new madecassic acid derivatives targeting erk cascade signaling. Front. Chem. 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.X.; Huang, J.Y.; Liu, C.M.; Zhang, J.Z.; Cheng, K.G. Synthesis of oleanolic acid/ursolic acid/glycyrrhetinic acid-hydrogen sulfide donor hybrids and their antitumor activity. Med. Chem. Res. 2019, 28, 1212–1222. [Google Scholar] [CrossRef]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of glycyrrhiza sp and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Baltina, L.A. Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr. Med. Chem. 2003, 10, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.K.; Iqbal, M.; Athar, M. Inhibitory effect of 18 beta-glycyrrhetinic acid on 12-o-tetradecanoyl phorbol-13-acetate-induced cutaneous oxidative stress and tumor promotion in mice. Redox Rep. 2005, 10, 151–157. [Google Scholar] [CrossRef]
- Kowsalya, R.; Vishwanathan, P.; Manoharan, S. Chemopreventive potential of 18beta-glycyrrhetinic acid: An active constituent of liquorice, in 7,12-dimethylbenz(a)anthracene induced hamster buccal pouch carcinogenesis. Pak. J. Biol. Sci. 2011, 14, 619–626. [Google Scholar] [CrossRef]
- Hasan, S.K.; Khan, R.; Ali, N.; Khan, A.Q.; Rehman, M.U.; Tahir, M.; Lateef, A.; Nafees, S.; Mehdi, S.J.; Rashid, S.; et al. 18-glycyrrhetinic acid alleviates 2-acetylaminofluorene-induced hepatotoxicity in wistar rats: Role in hyperproliferation, inflammation and oxidative stress. Hum. Exp. Toxicol. 2015, 34, 628–641. [Google Scholar] [CrossRef]
- Hasan, S.K.; Siddiqi, A.; Nafees, S.; Ali, N.; Rashid, S.; Ali, R.; Shahid, A.; Sultana, S. Chemopreventive effect of 18 beta-glycyrrhetinic acid via modulation of inflammatory markers and induction of apoptosis in human hepatoma cell line (hepg2). Mol. Cell. Biochem. 2016, 416, 169–177. [Google Scholar] [CrossRef]
- Cao, D.H.; Jiang, J.; Zhao, D.; Wu, M.H.; Zhang, H.J.; Zhou, T.Y.; Tsukamoto, T.; Oshima, M.; Wang, Q.; Cao, X.Y. The protective effects of 18 beta-glycyrrhetinic acid against inflammation microenvironment in gastric tumorigenesis targeting pge2-ep2 receptor-mediated arachidonic acid pathway. Eur. J. Inflamm. 2018, 16, 7. [Google Scholar] [CrossRef]
- Satomi, Y.; Nishino, H.; Shibata, S. Glycyrrhetinic acid and related compounds induce g1 arrest and apoptosis in human hepatocellular carcinoma hepg2. Anticancer Res. 2005, 25, 4043–4047. [Google Scholar]
- Zhu, J.; Chen, M.J.; Chen, N.; Ma, A.Z.; Zhu, C.Y.; Zhao, R.L.; Jiang, M.; Zhou, J.; Ye, L.H.; Fu, H.A.; et al. Glycyrrhetinic acid induces g1-phase cell cycle arrest in human non-small cell lung cancer cells through endoplasmic reticulum stress pathway. Int. J. Oncol. 2015, 46, 981–988. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Lee, M.S.; Han, E.S.; Lee, S.J. 18 beta-glycyrrhetinic acid induces apoptotic cell death in siha cells and exhibits a synergistic effect against antibiotic anti-cancer drug toxicity. Life Sci. 2008, 83, 481–489. [Google Scholar] [CrossRef]
- Sharma, G.; Kar, S.; Palit, S.; Das, P.K. 18 beta-glycyrrhetinic acid (concur) induces apoptosis through modulation of akt/foxo3a/bim pathway in human breast cancer mcf-7 cells. J. Cell. Physiol. 2012, 227, 1923–1931. [Google Scholar] [CrossRef]
- Wang, S.S.; Shen, Y.; Qiu, R.F.; Chen, Z.L.; Chen, Z.H.; Chen, W.B. 18 beta-glycyrrhetinic acid exhibits potent antitumor effects against colorectal cancer via inhibition of cell proliferation and migration. Int. J. Oncol. 2017, 51, 615–624. [Google Scholar] [CrossRef]
- Cai, Y.; Zhao, B.X.; Liang, Q.Y.; Zhang, Y.Q.; Cai, J.Y.; Li, G.F. The selective effect of glycyrrhizin and glycyrrhetinic acid on topoisomerase ii alpha and apoptosis in combination with etoposide on triple negative breast cancer mda-mb-231 cells. Eur. J. Pharmacol. 2017, 809, 87–97. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.H.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Kuang, P.H.; Zhao, W.X.; Su, W.X.; Zhang, Z.Q.; Zhang, L.; Liu, J.M.; Ren, G.L.; Yin, Z.Y.; Wang, X.M. 18 beta-glycyrrhetinic acid inhibits hepatocellular carcinoma development by reversing hepatic stellate cell-mediated immunosuppression in mice. Int. J. Cancer 2013, 132, 1831–1841. [Google Scholar] [CrossRef]
- Lai, Y.S.; Shen, L.H.; Zhang, Z.Z.; Liu, W.Q.; Zhang, Y.H.; Ji, H.; Tian, J. Synthesis and biological evaluation of furoxan-based nitric oxide-releasing derivatives of glycyrrhetinic acid as anti-hepatocellular carcinoma agents. Bio Org. Med. Chem. Lett. 2010, 20, 6416–6420. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Markov, A.V.; Logashenko, E.B.; Korchagina, D.V.; Zenkova, M.A.; Salakhutdinov, N.F.; Vlassov, V.V.; Tolstikov, G.A. Synthesis of novel 2-cyano substituted glycyrrhetinic acid derivatives as inhibitors of cancer cells growth and no production in lps-activated j-774 cells. Bio Org. Med. Chem. 2014, 22, 585–593. [Google Scholar] [CrossRef]
- Li, Y.; Feng, L.; Song, Z.F.; Li, H.B.; Huai, Q.Y. Synthesis and anticancer activities of glycyrrhetinic acid derivatives. Molecules 2016, 21, 20. [Google Scholar] [CrossRef]
- Guo, W.B.; Yan, M.M.; Xu, B.; Chu, F.H.; Wang, W.; Zhang, C.Z.; Jia, X.H.; Han, Y.T.; Xiang, H.J.; Zhang, Y.Z.; et al. Design, synthesis, and biological evaluation of the novel glycyrrhetinic acid-cinnamoyl hybrids as anti-tumor agents. Chem. Cent. J. 2016, 10, 11. [Google Scholar] [CrossRef]
- Xu, B.; Wu, G.R.; Zhang, X.Y.; Yan, M.M.; Zhao, R.; Xue, N.N.; Fang, K.; Wang, H.; Chen, M.; Guo, W.B.; et al. An overview of structurally modified glycyrrhetinic acid derivatives as antitumor agents. Molecules 2017, 22, 24. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Huai, X.D.; Zheng, Q.X.; Wang, W.; Li, H.J.; Huai, Q.Y. Design and preparation of derivatives of oleanolic and glycyrrhetinic acids with cytotoxic properties. Drug Des. Dev. Ther. 2018, 12, 1321–1336. [Google Scholar] [CrossRef]
- Lin, K.W.; Huang, A.M.; Hour, T.C.; Yang, S.C.; Pu, Y.S.; Lin, C.N. Beta 18b-glycyrrhetinic acid derivatives induced mitochondrial-mediated apoptosis through reactive oxygen species-mediated p53 activation in ntub1 cells. Bio Org. Med. Chem. 2011, 19, 4274–4285. [Google Scholar] [CrossRef]
- Drag-Zalesinska, M.; Kulbacka, J.; Saczko, J.; Wysocka, T.; Zabel, M.; Surowiak, P.; Drag, M. Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells. Bio Org. Med. Chem. Lett. 2009, 19, 4814–4817. [Google Scholar] [CrossRef]
- Schwarz, S.; Csuk, R. Synthesis and antitumour activity of glycyrrhetinic acid derivatives. Bio Org. Med. Chem. 2010, 18, 7458–7474. [Google Scholar] [CrossRef]
- Csuk, R.; Schwarz, S.; Siewert, B.; Kluge, R.; Strohl, D. Synthesis and cytotoxic activity of methyl glycyrrhetinate esterified with amino acids. Z. Fur Nat. Sect. B J. Chem. Sci. 2012, 67, 731–746. [Google Scholar] [CrossRef]
- Rao, G.; Kondaiah, P.; Singh, S.K.; Ravanan, P.; Sporn, M.B. Chemical modifications of natural triterpenes-glycyrrhetinic and boswellic acids: Evaluation of their biological activity. Tetrahedron 2008, 64, 11541–11548. [Google Scholar] [CrossRef]
- Maitraie, D.; Hung, C.F.; Tu, H.Y.; Liou, Y.T.; Wei, B.L.; Yang, S.C.; Wang, J.P.; Lin, C.N. Synthesis, anti-inflammatory, and antioxidant activities of 18 beta-glycyrrhetinic acid derivatives as chemical mediators and xanthine oxidase inhibitors. Bio Org. Med. Chem. 2009, 17, 2785–2792. [Google Scholar] [CrossRef]
- Sakano, K.; Ohshima, M. Microbial conversion of glycyrrhetinic acids .2. Microbial conversion of 18beta-glycyrrhetinic acid and 22-alpha-hydroxy-18beta-glycyrrhetinic acid by chainia-antibiotica. Agric. Biol. Chem. 1986, 50, 1239–1245. [Google Scholar] [CrossRef]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H.S. Bis(2-methoxyethyl)aminosulfur trifluoride: A new broad-spectrum deoxofluorinating agent with enhanced thermal stability. J. Org. Chem 1999, 64, 7048–7054. [Google Scholar] [CrossRef]
- High yield 11-de:Oxo:Glycyrrhetic Acid Prepn–by Reducing Glycyrrhetic Acid in Solvent Using Zinc and Hydrochloric Acid. JP59070638-A.; JP90024264-B, JP59070638-A. 21 April 1984.
- Csuk, R.; Schwarz, S.; Siewert, B.; Kluge, R.; Strohl, D. Synthesis and antitumor activity of ring a modified glycyrrhetinic acid derivatives. Eur. J. Med. Chem. 2011, 46, 5356–5369. [Google Scholar] [CrossRef]
- Liu, D.; Song, D.D.; Guo, G.; Wang, R.; Lv, J.L.; Jing, Y.K.; Zhao, L.X. The synthesis of 18 beta-glycyrrhetinic acid derivatives which have increased antiproliferative and apoptotic effects in leukemia cells. Bio Org. Med. Chem 2007, 15, 5432–5439. [Google Scholar] [CrossRef]
- Porchia, M.; Dolmella, A.; Gandin, V.; Marzano, C.; Pellei, M.; Peruzzo, V.; Refosco, F.; Santini, C.; Tisato, F. Neutral and charged phosphine/scorpionate copper(i) complexes: Effects of ligand assembly on their antiproliferative activity. Eur. J. Med. Chem. 2013, 59, 218–226. [Google Scholar] [CrossRef]
- Chu, F.H.; Xu, X.; Li, G.L.; Gu, S.; Xu, K.; Gong, Y.; Xu, B.; Wang, M.N.; Zhang, H.Z.; Zhang, Y.Z.; et al. Amino acid derivatives of ligustrazine-oleanolic acid as new cytotoxic agents. Molecules 2014, 19, 18215–18231. [Google Scholar] [CrossRef]
- Antunovic, M.; Kriznik, B.; Ulukaya, E.; Yilmaz, V.T.; Mihalic, K.C.; Madunic, J.; Marijanovic, I. Cytotoxic activity of novel palladium-based compounds on leukemia cell lines. Anti-Cancer Drugs 2015, 26, 180–186. [Google Scholar] [CrossRef]
- Rajic, Z.; Zorc, B.; Raic-Malic, S.; Ester, K.; Kralj, M.; Pavelic, K.; Balzarini, J.; De Clercq, E.; Mintas, M. Hydantoin derivatives of l- and d-amino acids: Synthesis and evaluation of their antiviral and antitumoral activity. Molecules 2006, 11, 837–848. [Google Scholar] [CrossRef]
- Goncalves, B.M.F.; Salvador, J.A.R.; Marin, S.; Cascante, M. Synthesis and biological evaluation of novel asiatic acid derivatives with anticancer activity. RSC Adv. 2016, 6, 3967–3985. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell line (IC50, µM) 1 | |
|---|---|---|
| A549 | HT-29 | |
| 1 | 110.5 ± 3.9 | 115.7 ± 1.6 |
| 5 | 59.4 ± 2.1 | 66.6 ± 3.2 |
| 6 | 31.6 ± 1.5 | 37.4 ± 1.0 |
| 7 | 26.2 ± 2.4 | 24.4 ± 1.7 |
| 10 | 52.2 ± 3.0 | 61.7 ± 1.4 |
| 11 | 33.7 ± 2.0 | 43.0 ± 1.5 |
| 12 | > 100 | > 100 |
| 13 | > 100 | > 100 |
| 14 | 33.7 ± 1.8 | 35.0 ± 1.7 |
| 15 | 26.4 ± 2.2 | 24.7 ± 0.9 |
| 16 | 24.4 ± 1.4 | 23.8 ± 0.3 |
| 17 | 14.8 ± 0.9 | 13.0 ± 0.5 |
| 18 | > 100 | > 100 |
| 19 | > 100 | > 100 |
| 20 | > 100 | > 100 |
| 21 | > 100 | > 100 |
| Cisplatin | 12.6 ± 0.8 [48] | 6.1 [49] |
| Compound | Cell line (IC50, µM) 1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Jurkat | MOLT-4 | MIAPaca2 | MCF7 | HeLa | A375 | HepG2 | BJ | |
| 1 | 105.6 ± 5.0 | 95.5 ± 3.9 | 101.6 ± 1.6 | 97.8 ± 3.9 | 107.2 ± 2.5 | 112.2 ± 2.6 | 125.1 ± 9.1 | 165.0 ± 7.1 |
| 6 | 11.9 ± 0.2 | 18.9 ± 1.6 | 28.2 ± 0.5 | 32.9 ± 1.6 | 34.5 ± 2.5 | 30.0 ± 1.5 | 30.6 ± 0.5 | N.D. |
| 7 | 11.7 ± 0.6 | 18.5 ± 0.9 | 24.9 ± 1.2 | 24.9 ± 0.9 | 25.7 ± 0.6 | 24.5 ± 1.0 | 24.8 ± 0.4 | N.D. |
| 14 | 13.3 ± 1.1 | 23.5 ± 0.8 | 32.5 ± 3.2 | 28.8 ± 0.7 | 34.2 ± 2.4 | 30.0 ± 2.2 | 34.7 ± 1.1 | N.D. |
| 15 | 12.5 ± 0.5 | 18.9 ± 1.6 | 20.2 ± 1.2 | 24.8 ± 1.3 | 22.2 ± 0.3 | 18.8 ± 1.1 | 25.4 ± 1.3 | N.D. |
| 16 | 9.6 ± 0.4 | 19.1 ± 1.3 | 22.6 ± 0.6 | 23.8 ± 1.6 | 19.1 ± 0.5 | 17.0 ± 1.1 | 25.7 ± 0.8 | N.D. |
| 17 | 6.1 ± 0.2 | 15.3 ± 0.7 | 11.8 ± 1.1 | 21.6 ± 0.6 | 13.0 ± 0.5 | 11.3 ± 0.4 | 16.0 ± 0.3 | > 100 |
| 18 | 46.4 ± 3.7 | 51.9 ± 2.5 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| 19 | 40.8 ± 2.7 | 49.0 ± 1.6 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| 20 | > 100 | > 100 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| 21 | > 100 | > 100 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Cisplatin | 1.9 [50] | 1.4 [50] | 5.0 ± 1.0 [51] | 19.1 ± 4.5 [52] | 2.3 ± 0.3 [52] | 3.1 ± 1.0 [48] | 2.9 [49] | 10.1 ± 2.0 [52] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alho, D.P.S.; Salvador, J.A.R.; Cascante, M.; Marin, S. Synthesis and Antiproliferative Activity of Novel A-Ring Cleaved Glycyrrhetinic Acid Derivatives. Molecules 2019, 24, 2938. https://doi.org/10.3390/molecules24162938
Alho DPS, Salvador JAR, Cascante M, Marin S. Synthesis and Antiproliferative Activity of Novel A-Ring Cleaved Glycyrrhetinic Acid Derivatives. Molecules. 2019; 24(16):2938. https://doi.org/10.3390/molecules24162938
Chicago/Turabian StyleAlho, Daniela P.S., Jorge A.R. Salvador, Marta Cascante, and Silvia Marin. 2019. "Synthesis and Antiproliferative Activity of Novel A-Ring Cleaved Glycyrrhetinic Acid Derivatives" Molecules 24, no. 16: 2938. https://doi.org/10.3390/molecules24162938
APA StyleAlho, D. P. S., Salvador, J. A. R., Cascante, M., & Marin, S. (2019). Synthesis and Antiproliferative Activity of Novel A-Ring Cleaved Glycyrrhetinic Acid Derivatives. Molecules, 24(16), 2938. https://doi.org/10.3390/molecules24162938