Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules


Abstract
:1. Introduction
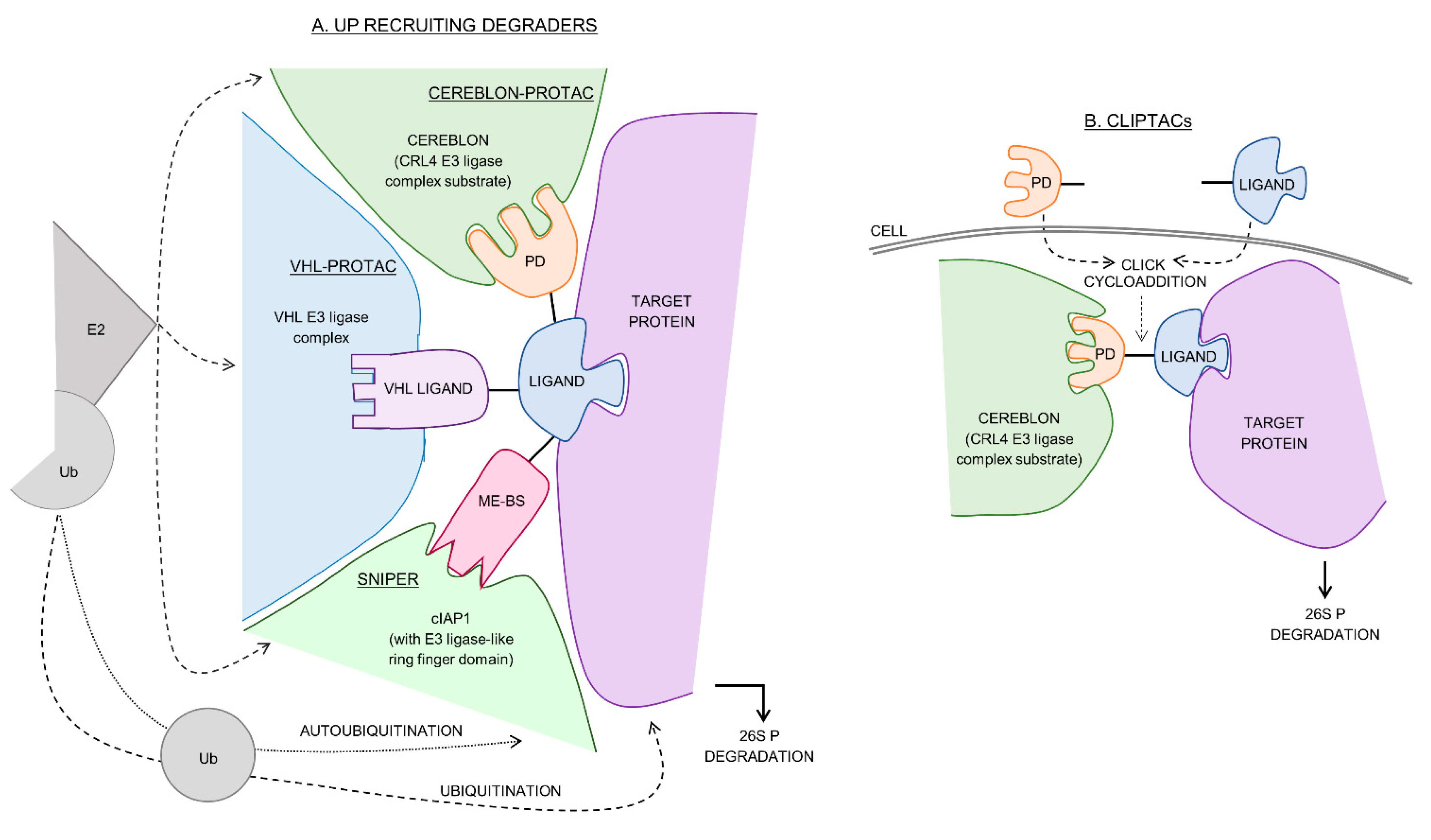
2. Cereblon-Recruiting PROTACs
3. VHL-Recruiting PROTACs
4. cIAP1 Recruiting SNIPERs
5. Other E3 Ligase Recruiting Protacs
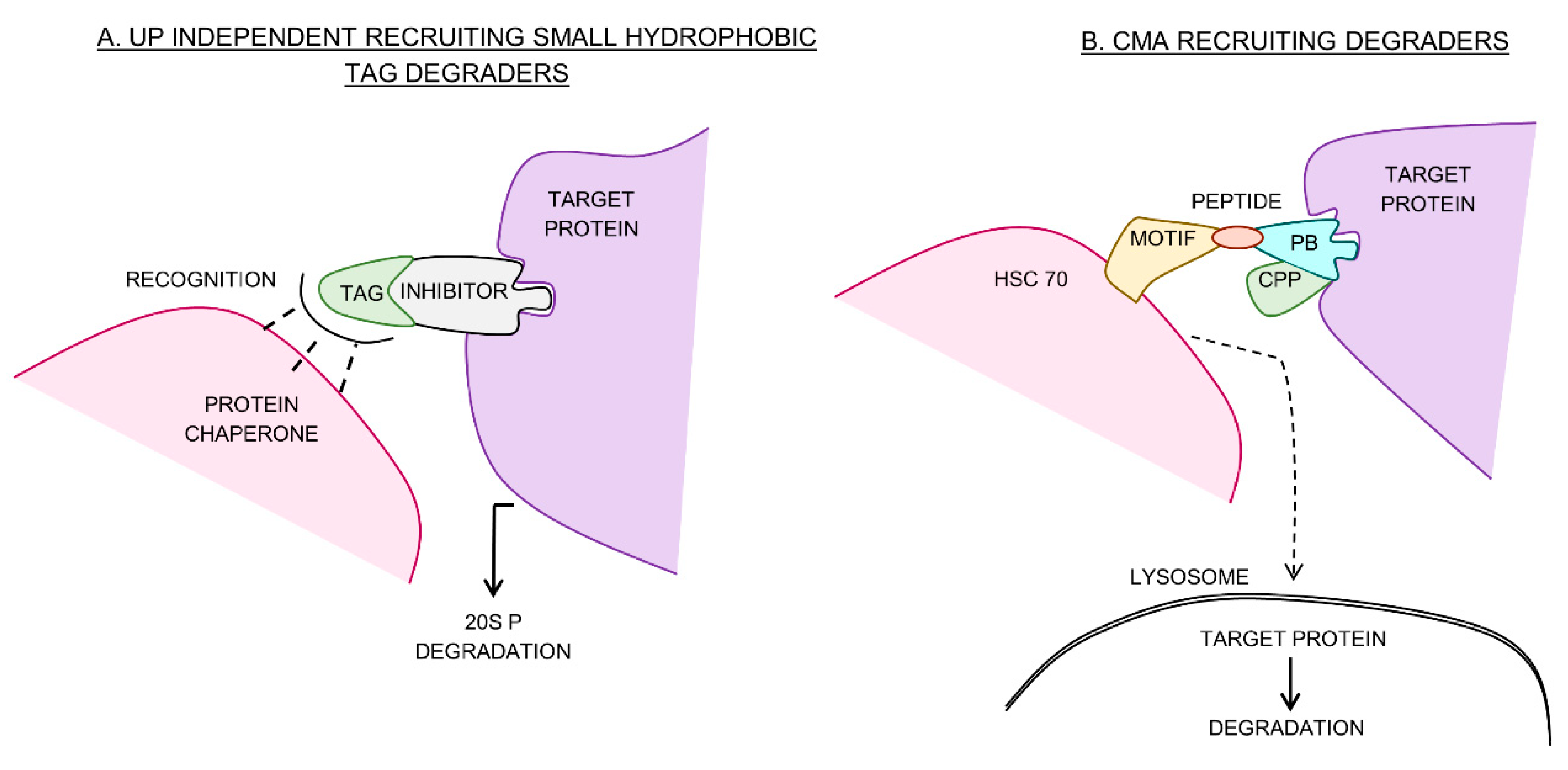
6. Ubiquitin Independent Small Hydrophobic Tag Degraders
7. Chaperone Mediated Autophagy-Recruiting Degraders
8. Conclusions
Funding
Conflicts of Interest
References
- Lapek, J.D.; Greninger, P.; Morris, R.; Amzallag, A.; Pruteanu-Malinici, I.; Benes, C.H.; Haas, W. Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. 2017, 35, 983–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Makley, L.N.; Gestwicki, J.E. Expanding the number of ‘druggable’ targets: Non-enzymes and protein–protein interactions. Chem. Biol. Drug Des. 2013, 81, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, S.-L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta. 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.; Gollapalli, D.R.; Hedstrom, L. Inhibitor mediated protein degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Xie, T.; Westover, K.D.; Ficarro, S.B.; Tae, H.S.; Gurbani, D.; Sim, T.; Marto, J.A.; Jänne, P.A.; Crews, C.M. Development of small molecules targeting the pseudokinase Her3. Bioorg. Med. Chem. Lett. 2015, 25, 3382–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Chemical protein degradation approach and its application to epigenetic targets. Chem. Rec. 2018, 18, 1681–1700. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2017, 25, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2017, 25, 67–77. [Google Scholar] [CrossRef]
- Huang, H.-T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.-H.; Ko, E.; Jang, J. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2017, 25, 88–99. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M. Could a common mechanism of protein degradation impairment underlie many neurodegenerative diseases? J. Exp. Neurosci. 2018, 12, 1179069518794675. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Go, A.; Jo, S.-H.; Park, S.J.; Jeon, Y.U.; Kim, J.E.; Lee, H.K.; Park, C.H.; Lee, C.-O.; Park, S.G.; et al. A novel cereblon modulator for targeted protein degradation. Eur. J. Med. Chem. 2019, 166, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET Proteins as targets for anti-cancer treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Selective target protein degradation via phthalimide conjugation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, B.; Yang, C.-Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.; Crew, A.; Shen, A.; Mill, C. Novel BET protein proteolysis targeting chimera (BET-PROTAC) exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm (MPN) secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2017, 32, 343–352. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.-Y.; Zhao, Y. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2017, 61, 462–481. [Google Scholar] [CrossRef]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.S.; Dastjerdi, S.; Wühr, M.; Dhe-Paganon, S.; Armstrong, S.A. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem. Int. Ed. 2017, 56, 5738–5743. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Fillmore, M.C.; Miah, A.H.; Chapman, T.D.; Maller, C.; Roberts, E.J.; Davis, L.C.; Lewis, D.E.; Galwey, N.W.; Waddington, K.E.; et al. Modulating PCAF/GCN5 immune cell function through a PROTAC approach. ACS Chem. Biol. 2018, 13, 2862–2867. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.r.; Swyter, S.r.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2017, 61, 482–491. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Chessum, N.E.A.; Sharp, S.Y.; Caldwell, J.J.; Pasqua, A.E.; Wilding, B.; Colombano, G.; Collins, I.; Ozer, B.; Richards, M.; Rowlands, M.; et al. Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem. 2018, 61, 918–933. [Google Scholar] [CrossRef]
- Raina, K.; Crews, C.M. Chemical inducers of targeted protein degradation. J. Biol. Chem. 2010, 285, 11057–11060. [Google Scholar] [CrossRef]
- Lee, H.; Puppala, D.; Choi, E.-Y.; Swanson, H.; Kim, K.-B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. ChemBioChem 2007, 8, 2058–2062. [Google Scholar] [CrossRef]
- Henning, R.K.; Varghese, J.O.; Das, S.; Nag, A.; Tang, G.; Tang, K.; Sutherland, A.M.; Heath, J.R. Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci. 2016, 22, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.-T.; Gao, N.; Li, Q.-Q.; Chen, P.-G.; Yang, X.-F.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Specific knockdown of endogenous Tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von hippel-lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Flanagan, J.; Qian, Y.; Gough, S.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5-04-18. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; et al. An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2018, 36, 381. [Google Scholar] [CrossRef]
- Müllard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019, 18, 237–239. [Google Scholar] [CrossRef]
- Bennett, C. New oral compound can target and degrade the estrogen receptor. Oncol. Times 2017, 39, 14–15. [Google Scholar] [CrossRef]
- Sekine, K.; Takubo, K.; Kikuchi, R.; Nishimoto, M.; Kitagawa, M.; Abe, F.; Nishikawa, K.; Tsuruo, T.; Naito, M. Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J. Biol. Chem. 2008, 283, 8961–8968. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin− ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Okuhira, K.; Ohoka, N.; Sai, K.; Nishimaki-Mogami, T.; Itoh, Y.; Ishikawa, M.; Hashimoto, Y.; Naito, M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 2011, 585, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg. Med. Chem. Lett. 2011, 19, 6768–6778. [Google Scholar] [CrossRef]
- Demizu, Y.; Okuhira, K.; Motoi, H.; Ohno, A.; Shoda, T.; Fukuhara, K.; Okuda, H.; Naito, M.; Kurihara, M. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 2012, 22, 1793–1796. [Google Scholar] [CrossRef]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R. In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef]
- Ohoka, N.; Nagai, K.; Hattori, T.; Okuhira, K.; Shibata, N.; Cho, N.; Naito, M. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 2014, 5, e1513. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of small molecules that induce degradation of huntingtin. Angew. Chem. Int. Ed. 2017, 129, 11688–11691. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett. 2018, 28, 707–710. [Google Scholar] [CrossRef]
- Zhong, D.; Ru, Y.; Wang, Q.; Zhang, J.; Zhang, J.; Wei, J.; Wu, J.; Yao, L.; Li, X.; Li, X. Chimeric ubiquitin ligases inhibit non-small cell lung cancer via negative modulation of EGFR signaling. Cancer Lett. 2015, 359, 57–64. [Google Scholar] [CrossRef]
- Ottis, P.; Toure, M.; Cromm, P.M.; Ko, E.; Gustafson, J.L.; Crews, C.M. Assessing different E3 ligases for small molecule induced protein ubiquitination and degradation. ACS Chem. Biol. 2017, 12, 2570–2578. [Google Scholar] [CrossRef]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-recruiting PROTAC offers superior, synergistic anti-proliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-linked ligands induce degradation by localizing target proteins to the 20S proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef]
- Wang, L.; Guillen, V.S.; Sharma, N.; Flessa, K.; Min, J.; Carlson, K.E.; Toy, W.; Braqi, S.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; et al. New class of selective estrogen receptor degraders (SERDs): Expanding the toolbox of PROTAC degrons. ACS Med. Chem. Let. 2018, 9, 803–808. [Google Scholar] [CrossRef]
- Gao, N.; Chu, T.-T.; Li, Q.-Q.; Lim, Y.-J.; Qiu, T.; Ma, M.-R.; Hu, Z.-W.; Yang, X.-F.; Chen, Y.-X.; Zhao, Y.-F.; et al. Hydrophobic tagging-mediated degradation of Alzheimer’s disease related Tau. RSC Adv. 2017, 7, 40362–40366. [Google Scholar] [CrossRef]
- Gao, N.; Huang, Y.-P.; Chu, T.-T.; Li, Q.-Q.; Zhou, B.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. TDP-43 specific reduction induced by di-hydrophobic tags conjugated peptides. Bioorg. Chem. 2019, 84, 254–259. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. PROTACs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.G.; Chang, L.; Gestwicki, J.E. Heat shock protein 70 (Hsp70) as an emerging drug target. J. Med. Chem. 2010, 53, 4585–4602. [Google Scholar] [CrossRef]
- Müller, M.P.; Rauh, D. Try me: Promiscuous inhibitors still allow for selective targeted protein degradation. Cell Chem. Biol. 2018, 25, 4–6. [Google Scholar] [CrossRef]
- Kaneko, M.; Iwase, I.; Yamasaki, Y.; Takai, T.; Wu, Y.; Kanemoto, S.; Matsuhisa, K.; Asada, R.; Okuma, Y.; Watanabe, T.; et al. Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci. Rep. 2016, 6, 30955. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, W.; Xiao, Z.; Gan, H.; Lin, X.; Liao, S.; Han, C. Mining and characterization of ubiquitin E3 ligases expressed in the mouse testis. BMC Genom. 2012, 13, 495. [Google Scholar] [CrossRef]
- Yamada, T.; Yang, Y.; Bonni, A. Spatial organization of ubiquitin ligase pathways orchestrates neuronal connectivity. Trends Neurosci. 2013, 36, 218–226. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Type of Degrader | Degrader Name | Degrader Target | Disease | DC50 a (nM) | Reference |
|---|---|---|---|---|---|
| Cereblon-recruiting PROTACs: | dBET1 | BRD4 | Cancer (MM, AML) | 430.0 | Winter, et al. [26] |
| TD-428 | BRD4 | Cancer (MM) | 32.0 × 10−2 | Kim, et al. [24] | |
| ARV-825 | BRD4 | Cancer (Burkitt lymphoma) | 1.0 | Lu, et al. [28] | |
| BETd-246 | BRD4 | Cancer (breast) | 10.0 | Bai, et al. [27] | |
| Compound 21 | BRD4 | Cancer (MM, AML) | 3.7 × 10−2 | Zhou, et al. [31] | |
| Compound 23 | BRD4 | 5.1 × 10−2 | |||
| dBRD9 | BRD9 | Cancer (AML) | 104.0 | Remillard, et al. [32] | |
| GSK983 | PCAF | Anti-inflammatory diseases | 1.5 | Bassi, et al. [33] | |
| GCN5 | 3.0 | ||||
| DAS-6-2-2-6-CRBN | cAbl | Cancer (chronic myelogenous leukemia) | 25.0 | Lai, et al. [34] | |
| Bcr-Abl | |||||
| TL12-186 | CDK2 | Cancer, rheumatoid arthritis, and idiopathic pulmonary fibrosis | 73.0 | Huang, et al. [18] | |
| CDK9 | 55.0 | ||||
| CRBN-PROTAC 2 | p38δ | Cancer and Diabetes | 27.0 | Bondeson, et al. [16] | |
| MT-802 | BTK | Chronic lymphocytic leukemia | 9.1 | Buhimschi, et al. [35] | |
| pomalidomide-Vorinostat | HDAC6 | Cancer (AML, ovarian, hepatocellular carcinomas) | 32.0 | Yang, et al. [39] | |
| VHL-recruiting PROTACs: | MZ1 | BRD4 | Cancer (NSCLC) | 1000.0 | Zhong, et al. [65] |
| ARV-771 | BRD4 | Cancer (castration-resistant prostate cancer) | 5.0 | Raina, et al. [46] | |
| VZ185 | BRD9 | Cancer (cervical, NSCLC) | 4.0 | Zoppi, et al. [47] | |
| BRD7 | 34.0 | ||||
| ACBI1 | SMARCA2 | Cancer (AML) | 6.0 | Farnaby, et al. [48] | |
| SMARCA4 | 11.0 | ||||
| PBRM1 | 32.0 | ||||
| PROTAC_RIPK2 | RIPK2 | Auto-inflammatory diseases (Blau syndrome, early-onset sarcoidosis) | 1.4 | Bondeson, et al. [49] | |
| PROTAC_ERRα | ERRα | Cancer (breast) | 100.0 | ||
| VHL-PROTAC 1 | p38α | Cancer | 210.0 | Bondeson, et al. [16] | |
| SJFα | p38α | 7.2 | Smith, et al. [19] | ||
| SJFδ | p38δ | Cancer and diabetes | 46.2 | ||
| VHL-lapatinib | EGFR | Cancer (glioblastoma multiforme, NSCLC) | 39.2 | Burslem, et al. [17] | |
| VHL-gefitinib | 11.7 | ||||
| VHL-afatinib | 215.8 | ||||
| VHL-lapatinib | HER2 | 102.0 | |||
| VHL-Foretinib | c-MET | 66.7 | |||
| PROTAC-3 | Fak | Cancer (malignant pleural mesothelioma, ovarian) | 3.0 | Cromm, et al. [50] | |
| Nondisclosed recruiting PROTACs: | ARV-471 | ERα | Cancer (breast) | 2.0 | Flanagan, et al. [52] |
| AR PROTAC | AR | Cancer (prostate) | 1.0 | Neklesa, et al. [53] | |
| SNIPERs: | SNIPER(CRABP-I) | CRABP-I | ~10,000.0 b | Itoh, et al. [57] | |
| SNIPER(ERα)-87 | ERα (MCF-7 cells) | Cancer (breast) | 3.0 | Ohoka, et al. [61] | |
| ERα (T47D cells) | Cancer (breast) | 9.6 | |||
| SNIPER(TACC3) | TACC3 | Cancer (ovarian, breast, squamous cell carcinoma, lymphoma) | ~10,000.0 b | Ohoka, et al. [62] | |
| SNIPER(ABL)-38 | cABL/BCR-ABL | Cancer (chronic myelogenous leukemia, MM, AML) | 30.0 | Ohoka, et al. [61] | |
| SNIPER(BRD4)-1 | BRD4 | 10.0 | |||
| SNIPER(PDE4)-9 | PDE4 | 10.0 b | |||
| MDM2-recruiting PROTACs: | A1874 | BRD4 | Cancer (MM, AML) | 32.0 | Hines, et al. [69] |
| UP independent HyT degraders: | SARD279 | AR | Cancer (prostate) | 1000.0 | Gustafson, et al. [11] |
| SARD033 | 2000.0 | ||||
| CMA-recruiting degraders: | TAT-GluN2Bct-PP | DAPK1 | Neuroprotectivity (stroke) | ~50,000.0 b | Fan, et al. [79] |
| Strategy | Advantages | Disadvantages | |
|---|---|---|---|
| Ub Dependent | Cereblon-recruiting PROTACs | > Increases specificity of promiscuous inhibitors > Most efficient degraders (DC50 in pM range) | > Possible off-target degradation (e.g., IKZF) |
| CLIPTACs | > More drug-like scaffold | > Small target protein test group | |
| VHL-recruiting PROTACs | > Increases specificity of promiscuous inhibitors > No off-target degradation | > Lack efficiency when compared to cereblon PROTACs (DC50 nM range) | |
| SNIPERs | > First small molecule degrader to target a neurodegenerative disease-related protein | > Autoubiquitination and simultaneous degradation of cIAP a > Lack efficiency when compared to cereblon PROTACs (DC50 in nM range) | |
| Ub Independent | HyT degraders | > More drug-like scaffold | > Lack efficiency (DC50 in mM range) |
| CMA degraders | > Could potentially be used to treat diseases where the UP cascade is dysfunctional | > Peptidic structure | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delport, A.; Hewer, R. Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules. Molecules 2019, 24, 3272. https://doi.org/10.3390/molecules24183272
Delport A, Hewer R. Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules. Molecules. 2019; 24(18):3272. https://doi.org/10.3390/molecules24183272
Chicago/Turabian StyleDelport, Alexandré, and Raymond Hewer. 2019. "Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules" Molecules 24, no. 18: 3272. https://doi.org/10.3390/molecules24183272
APA StyleDelport, A., & Hewer, R. (2019). Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules. Molecules, 24(18), 3272. https://doi.org/10.3390/molecules24183272