Effects of High Pressure Modification on Conformation and Digestibility Properties of Oyster Protein

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
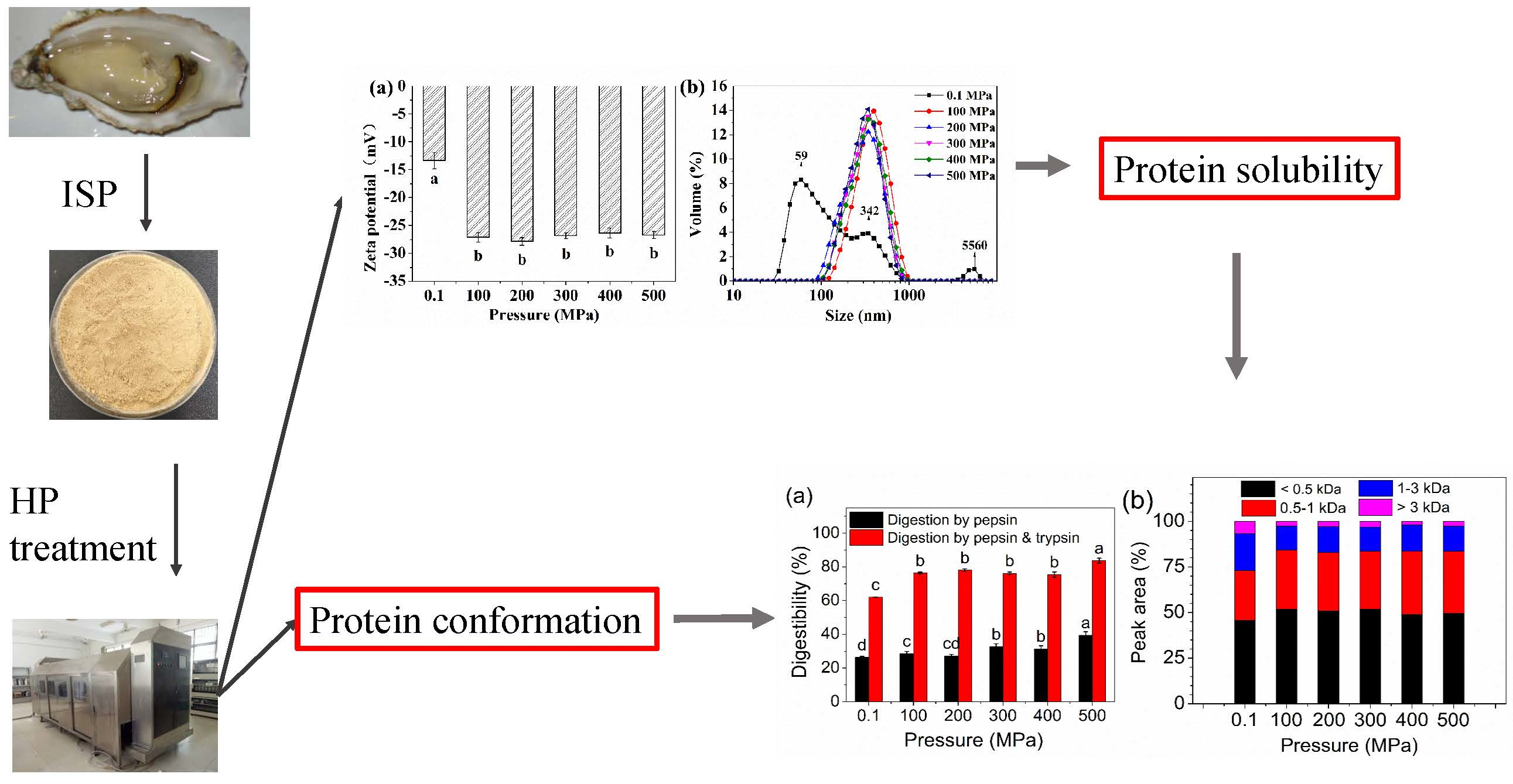
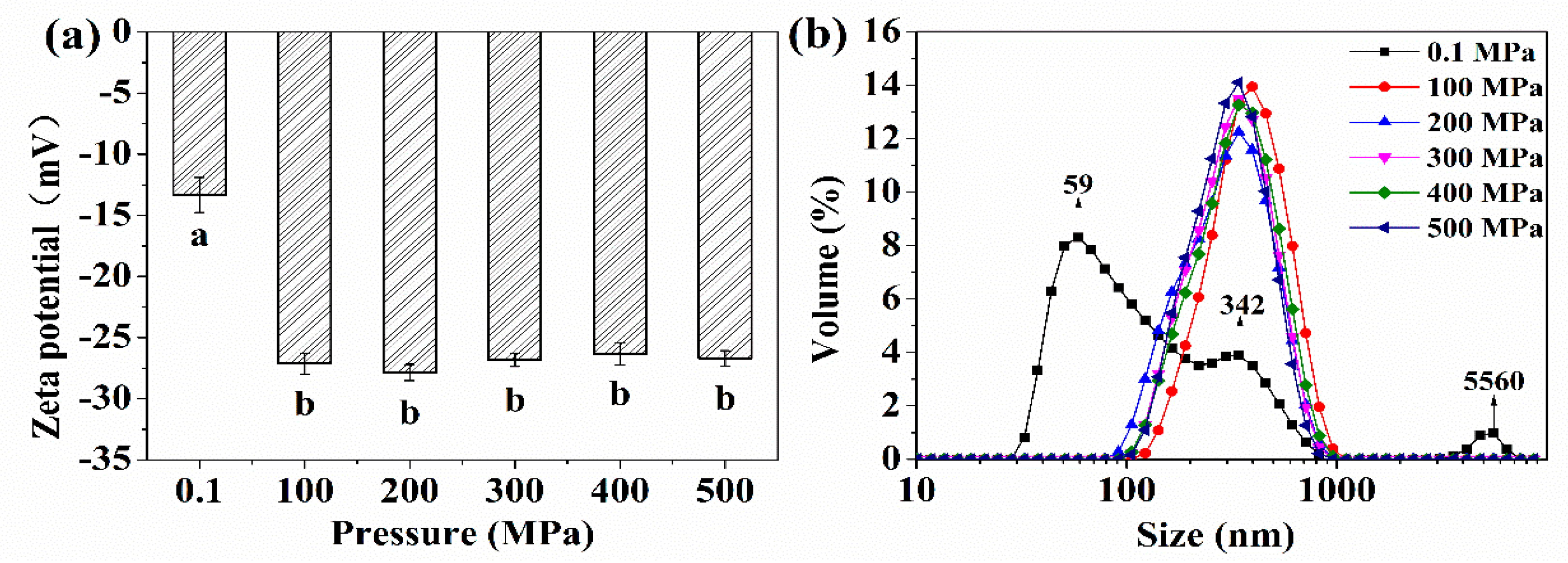
2.1. Zeta Potential, Particle Size Distribution, and Solubility
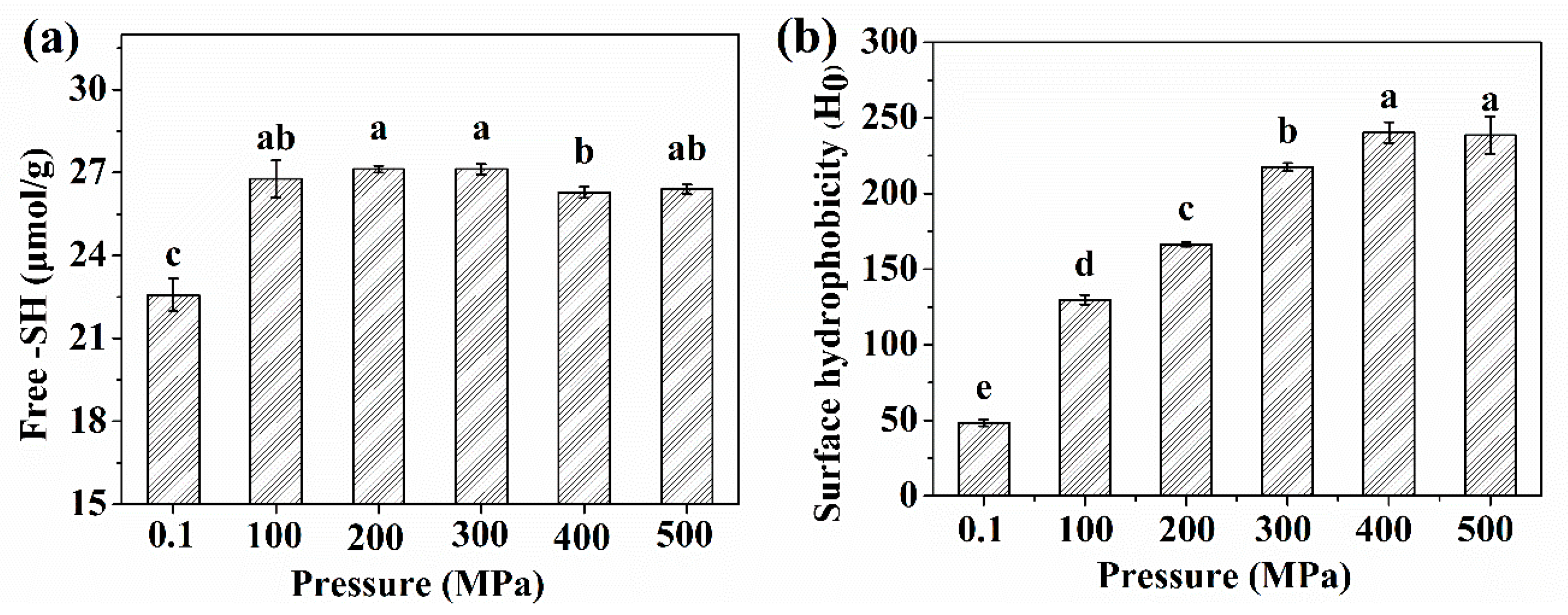
2.2. The -SH Groups and Surface Hydrophobicity
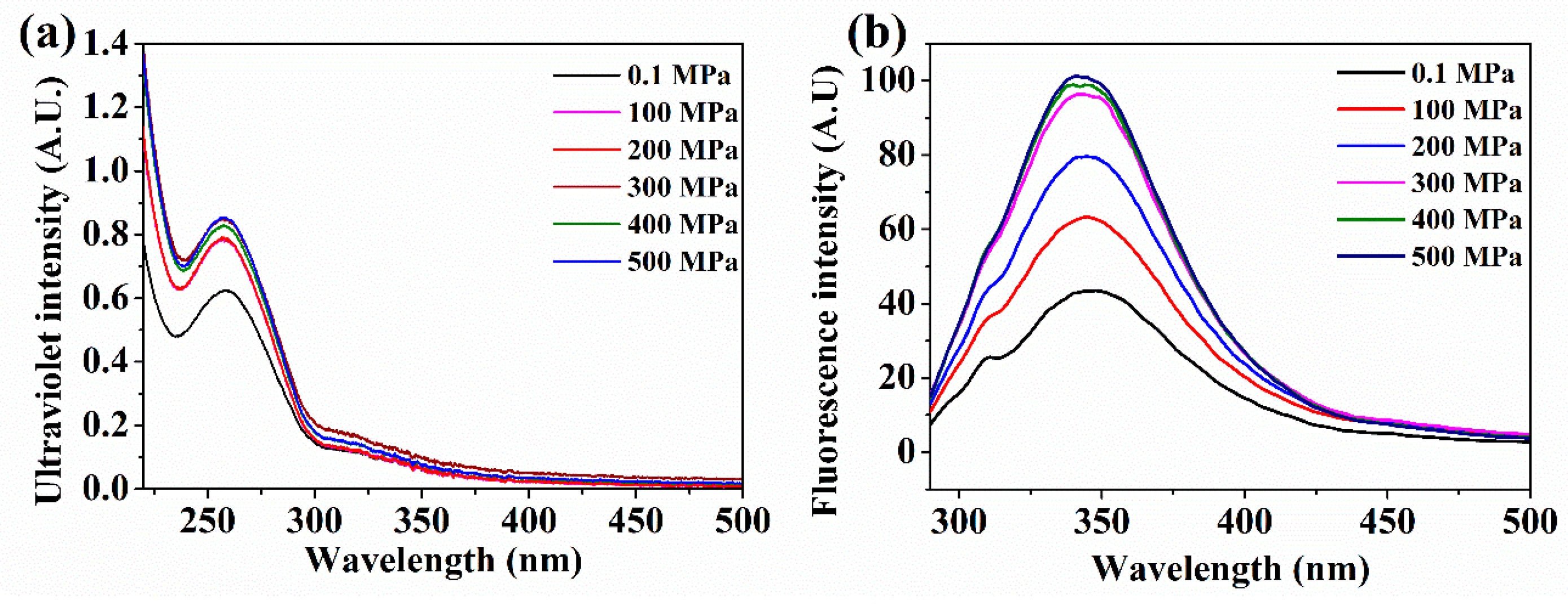
2.3. UV–Vis Spectrum and Intrinsic Fluorescence Spectroscopy
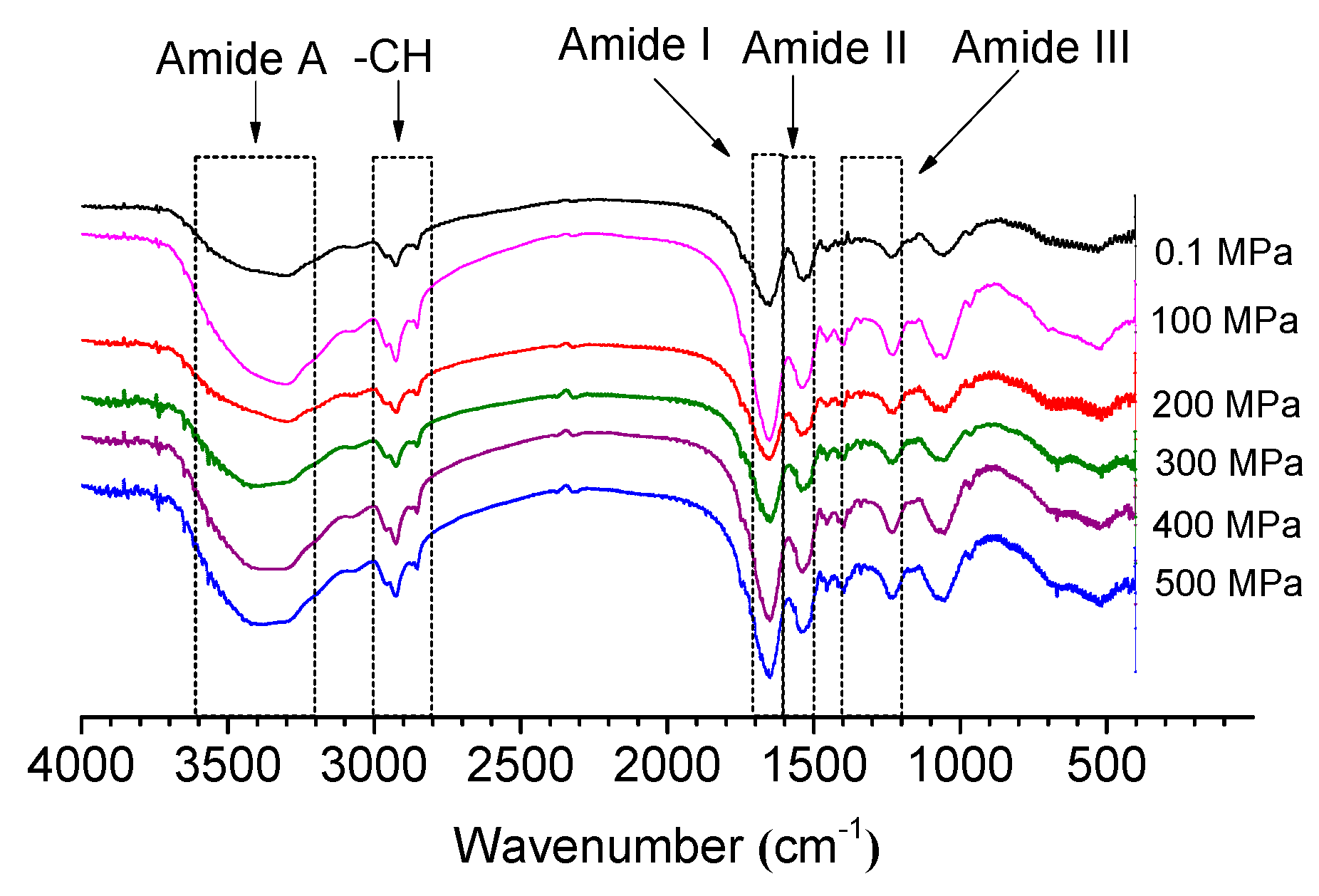
2.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.4.1. Influence of HP Treatment on Amide I Bands and Amide II Bands
2.4.2. Influence on Amide A and –CH under HP Treatment
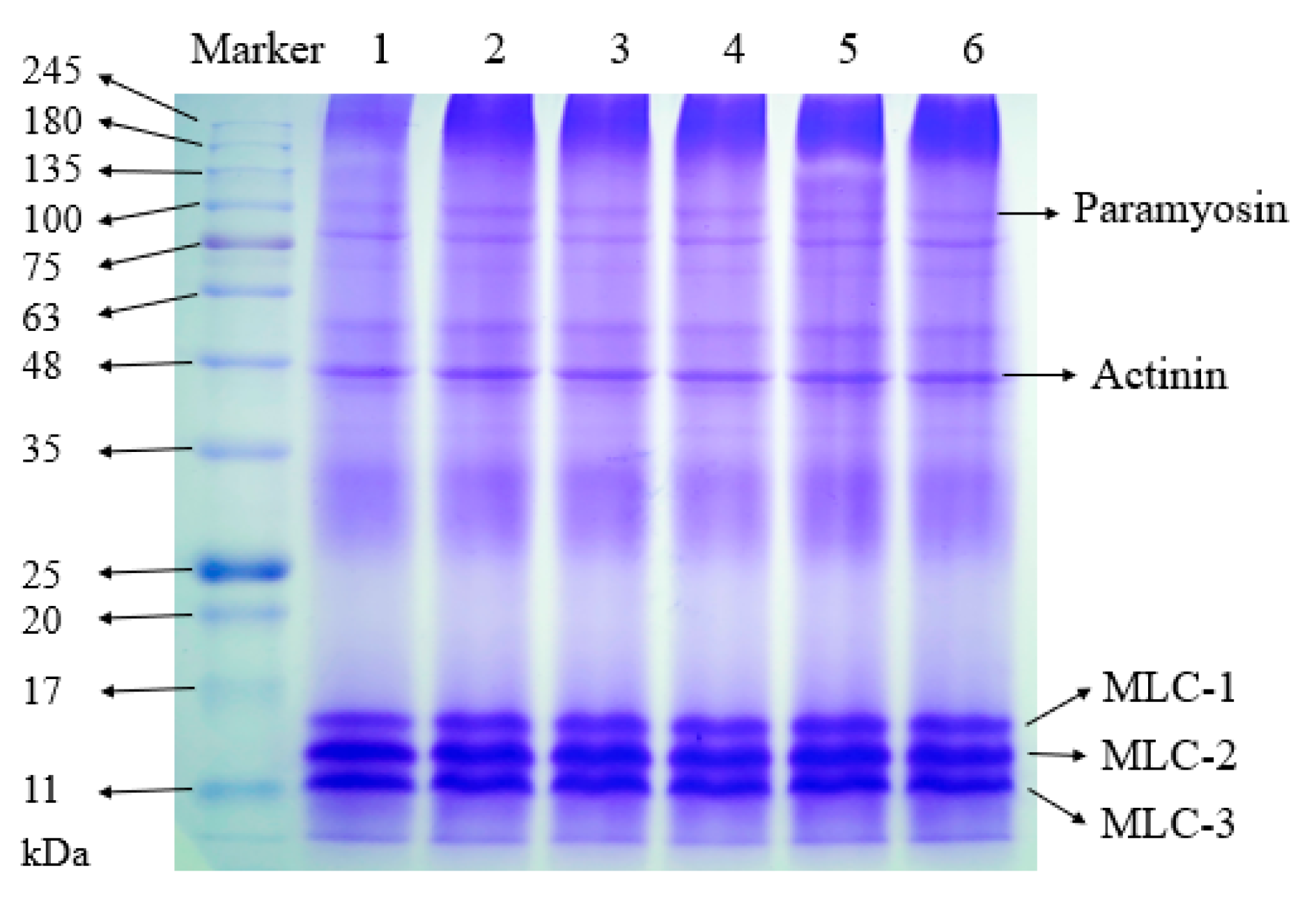
2.5. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
2.6. Microstructure Analysis
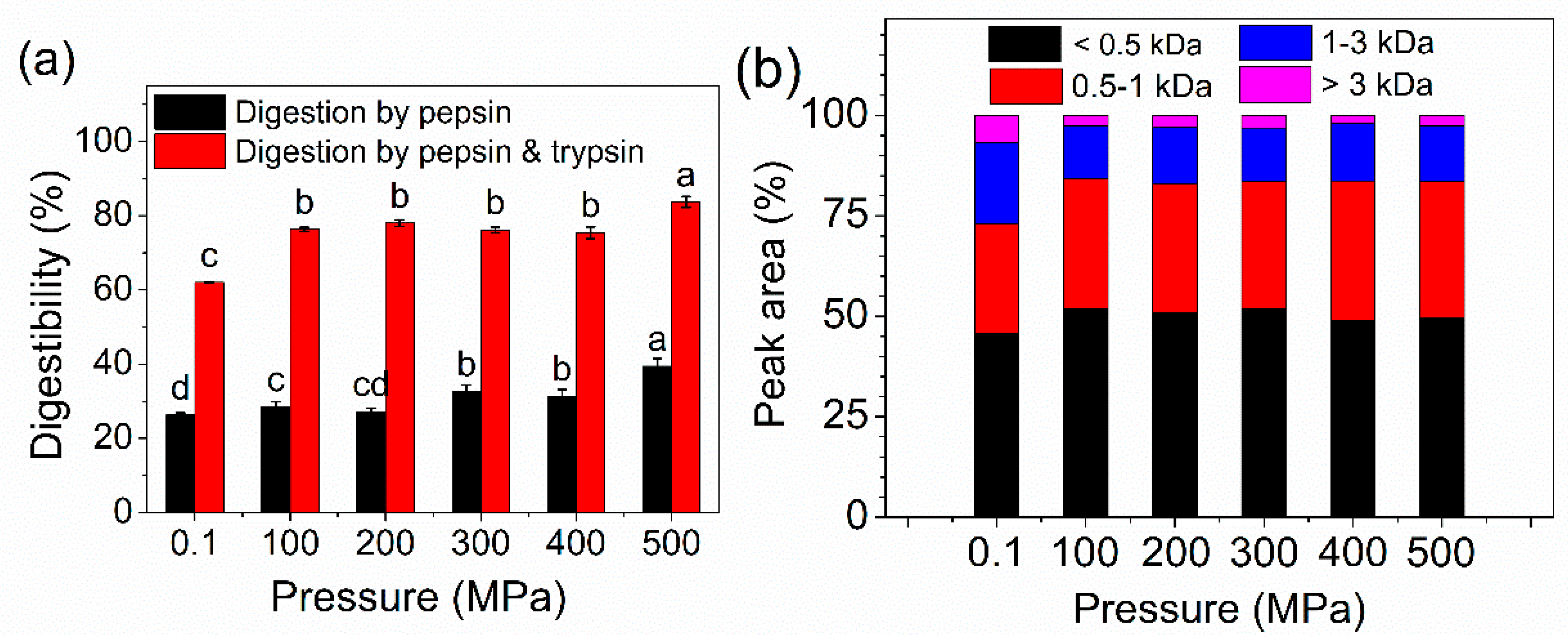
2.7. In Vitro Digestibility and MW Distribution of Peptides
3. Materials and Methods
3.1. Materials
3.2. Preparation of OP
3.3. High-Pressure Treatment
3.4. Measurement of Protein Solubility
3.5. Measurement of Particle Size Distribution and Zeta Potential
3.6. Measurement of Sulfhydryl Group Content
3.7. Measurement of Surface Hydrophobicity
3.8. UV–Vis Spectra
3.9. Intrinsic Fluorescence Spectroscopy
3.10. Fourier Transform Infrared Spectroscopy (FT-IR)
3.11. SDS-PAGE
3.12. Scanning Electron Microscopy (SEM) Observations
3.13. In Vitro Digestibility
3.14. Estimation of Peptide MW Distribution
3.15. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, X.; Yu, H.; Xing, R.; Liu, S.; Chen, X.; Li, P. Effect and mechanism of oyster hydrolytic peptides on spatial learning and memory in mice. RSC Adv. 2018, 8, 6125–6135. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, W.; He, Y.; Ren, D.; Kow, F.; Song, L.; Yu, X. Novel antioxidative peptides from the protein hydrolysate of oysters (Crassostrea talienwhanensis). Food Chem. 2014, 145, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, R.; Wang, Z. Nutritional components and proteolysis of oyster meat. J. Fish. China 2003, 27, 163–168. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Zhang, C.; Hao, J.; Zhang, J.; Zhang, J. The separation and composition of Oyster protein. Food Ferment. Ind. 2013, 39, 195–199. [Google Scholar] [CrossRef]
- Xie, X.; Zhao, W.; Sun, Q.; Yang, R. Separation of Glycogen and Protein from Oyster with Organic Acid. J. Food Sci. Technol. 2016. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, J.; Zhou, C.; Zhang, C.; Qin, X.; Ji, H.; Huang, M. Extraction of oyster protein by alkali solution acid precipitation and its amino acid and protein composition analysis. J. Chin. Inst. Food Sci. Technol. 2014, 14, 230–235. [Google Scholar] [CrossRef]
- Zhao, Z.-K.; Mu, T.-H.; Zhang, M.; Richel, A. Chemical forces, structure, and gelation properties of sweet potato protein as affected by pH and high hydrostatic pressure. Food Bioprocess Technol. 2018, 11, 1719–1732. [Google Scholar] [CrossRef]
- Wang, X.-S.; Tang, C.-H.; Li, B.-S.; Yang, X.-Q.; Li, L.; Ma, C.-Y. Effects of high-pressure treatment on some physicochemical and functional properties of soy protein isolates. Food Hydrocoll. 2008, 22, 560–567. [Google Scholar] [CrossRef]
- Puppo, C.; Chapleau, N.; Speroni, F.; de Lamballerie-Anton, M.; Michel, F.; Añón, C.; Anton, M. Physicochemical modifications of high-pressure-treated soybean protein isolates. J. Agric. Food Chem. 2004, 52, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- He, R.; He, H.-Y.; Chao, D.; Ju, X.; Aluko, R. Effects of high pressure and heat treatments on physicochemical and gelation properties of rapeseed protein isolate. Food Bioprocess Technol. 2014, 7, 1344–1353. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, R.; Xu, X.; Zhou, G.; Liu, D. Structural modification by high-pressure homogenization for improved functional properties of freeze-dried myofibrillar proteins powder. Food Res. Int. 2017, 100, 193–200. [Google Scholar] [CrossRef] [PubMed]
- De Maria, S.; Ferrari, G.; Maresca, P. Effects of high hydrostatic pressure on the conformational structure and the functional properties of bovine serum albumin. Innov. Food Sci. Emerg. Technol. 2016, 33, 67–75. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Zhou, P.; Zhang, X.; Wang, J. Effects of high pressure modification on conformation and gelation properties of myofibrillar protein. Food Chem. 2017, 217, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.M.; Lin, S.L.; Ramaswamy, H.S.; Yu, Y.; Zhang, Q.T. Enhancement of functional properties of rice bran proteins by high pressure treatment and their correlation with surface hydrophobicity. Food Bioprocess Technol. 2017, 10, 317–327. [Google Scholar] [CrossRef]
- Yu, C.; Cha, Y.; Wu, F.; Xu, X.; Du, M. Effects of limited hydrolysis and high-pressure homogenization on functional properties of oyster protein isolates. Molecules 2018, 23, 729. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, X.; Zhou, G. Potential of high pressure homogenization to solubilize chicken breast myofibrillar proteins in water. Innov. Food Sci. Emerg. Technol. 2016, 33, 170–179. [Google Scholar] [CrossRef]
- Keerati-u-rai, M.; Corredig, M. Effect of dynamic high pressure homogenization on the aggregation state of soy protein. J. Agric. Food Chem. 2009, 57, 3556–3562. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chi, Y.-J.; Xu, W. Comparisons on the functional properties and antioxidant activity of spray-dried and freeze-dried egg white protein hydrolysate. Food Bioprocess Technol. 2012, 5, 2342–2352. [Google Scholar] [CrossRef]
- Tian, Y.; Du, J.-B. Effect of disulfide bond and mercapto-group on structure and function of protein and analytical method. J. Appl. Clin. Pediatr. 2007, 22, 1499. [Google Scholar] [CrossRef]
- Condés, M.C.; Speroni, F.; Mauri, A.; Añón, M.C. Physicochemical and structural properties of amaranth protein isolates treated with high pressure. Innov. Food Sci. Emerg. Technol. 2012, 14, 11–17. [Google Scholar] [CrossRef]
- Shen, X.; Fang, T.; Gao, F.; Guo, M. Effects of ultrasound treatment on physicochemical and emulsifying properties of whey proteins pre-and post-thermal aggregation. Food Hydrocoll. 2017, 63, 668–676. [Google Scholar] [CrossRef]
- Condes, M.C.; Anon, M.C.; Mauri, A.N. Amaranth protein films prepared with high-pressure treated proteins. J. Food Eng. 2015, 166, 38–44. [Google Scholar] [CrossRef]
- Lakemond, C.M.M.; de Jongh, H.H.J.; Hessing, M.; Gruppen, H.; Voragen, A.G.J. Soy glycinin: Influence of pH and ionic strength on solubility and molecular structure at ambient temperatures. J. Agric. Food Chem. 2000, 48, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Benelhadj, S.; Gharsallaoui, A.; Degraeve, P.; Attia, H.; Ghorbel, D. Effect of pH on the functional properties of Arthrospira (Spirulina) platensis protein isolate. Food Chem. 2016, 194, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, G.; Zeng, H.; Chen, L. Effects of high pressure homogenization on faba bean protein aggregation in relation to solubility and interfacial properties. Food Hydrocoll. 2018, 83, 275–286. [Google Scholar] [CrossRef]
- Yin, S.-W.; Tang, C.-H.; Wen, Q.-B.; Yang, X.-Q.; Li, L. Functional properties and in vitro trypsin digestibility of red kidney bean (Phaseolus vulgaris L.) protein isolate: Effect of high-pressure treatment. Food Chem. 2008, 110, 938–945. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.-Y.; Mu, T.-H.; Zhang, M.; Sun, H.-N.; Chen, J.-W.; Yu, M. Effects of pH and high hydrostatic pressure on the structural and rheological properties of sugar beet pectin. Food Hydrocoll. 2016, 60, 161–169. [Google Scholar] [CrossRef]
- Siddique, M.A.; Maresca, P.; Pataro, G.; Ferrari, G. Effect of pulsed light treatment on structural and functional properties of whey protein isolate. Food Res. Int. 2016, 87, 189–196. [Google Scholar] [CrossRef]
- Savadkoohi, S.; Bannikova, A.; Mantri, N.; Kasapis, S. Structural properties of condensed ovalbumin systems following application of high pressure. Food Hydrocoll. 2016, 53, 104–114. [Google Scholar] [CrossRef]
- Fan, M.; Hu, T.; Zhao, S.; Xiong, S.; Xie, J.; Huang, Q. Gel characteristics and microstructure of fish myofibrillar protein/cassava starch composites. Food Chem. 2017, 218, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Zhang, T.; Wang, Q.; Chen, Y.; Su, X. Isolation, Purification and Characterization of Glycoprotein from Oyster Juice. J. Chin. Inst. Food Sci. Technol. 2013, 13, 179–184. [Google Scholar] [CrossRef]
- You, L.; Li, Y.; Zhao, H.; Regenstein, J.; Zhao, M.; Ren, J. Purification and Characterization of an Antioxidant Protein from Pearl Oyster (Pinctada fucata martensii). J. Aquat. Food Prod. Technol. 2015, 24, 661–671. [Google Scholar] [CrossRef]
- Xiong, Y.L. Myofibrillar protein from different muscle-fiber types: Implications of biochemical and functional-properties in meat processing. Crit. Rev. Food Sci. Nutr. 1994, 34, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Shima, J.; Saeki, H. Solubility improvement of shellfish muscle proteins by reaction with glucose and its soluble state in low-ionic-strength medium. J. Agric. Food Chem. 2002, 50, 4327–4332. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.W.; Zhou, Y.H.; Liang, H.Q.; Hui, Y.; Zhou, H.Z. Study on protein purification and enzymatic hydrolysis of oyster. Mod. Food Sci. Technol. 2006. [Google Scholar] [CrossRef]
- Chen, J.; Liang, R.H.; Liu, W.; Liu, C.M.; Li, T.; Tu, Z.C.; Wan, J. Degradation of high-methoxyl pectin by dynamic high pressure microfluidization and its mechanism. Food Hydrocoll. 2012, 28, 121–129. [Google Scholar] [CrossRef]
- Sun, C.; Dai, L.; Liu, F.; Gao, Y. Simultaneous treatment of heat and high pressure homogenization of zein in ethanol–water solution: Physical, structural, thermal and morphological characteristics. Innov. Food Sci. Emerg. Technol. 2016, 34, 161–170. [Google Scholar] [CrossRef]
- Dufour, E.; Herve, G.; Haertle, T. Hydrolysis of beta-lactoglobulin by thermolysin and pepsin under high hydrostatic-pressure. Biopolymers 1995, 35, 475–483. [Google Scholar] [CrossRef]
- Xu, K.; Shi, D.; Hu, Y.; Qiu, R.; Zhao, C. Hydrolysis of hordein by different proteinase. Food Sci. Technol. 2010, 35, 13–28. [Google Scholar] [CrossRef]
- Chun, J.-Y.; Jo, Y.-J.; Min, S.-G.; Hong, G.-P. Effect of high pressure on the porcine placenral hydrolyzing activity of pepsin, trypsin and chymotrypsin. Korean J. Food Sci. Anim. Resour. 2014, 34, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Makoto, S.; Ok, S.D. Food-derived peptides and intestinal functions. Curr. Pharm. Des. 2010, 13, 885–895. [Google Scholar]
- Xie, N.; Liu, S.; Wang, C.; Li, B. Stability of casein antioxidant peptide fractions during in vitro digestion/Caco-2 cell model: Characteristics of the resistant peptides. Eur. Food Res. Technol. 2014, 239, 577–586. [Google Scholar] [CrossRef]
- Cui, C.; Zhao, M.; Yuan, B.; Zhang, Y.; Ren, J. Effect of pH and pepsin limited hydrolysis on the structure and functional properties of soybean protein hydrolysates. J. Food Sci. 2013, 78, C1871–C1877. [Google Scholar] [CrossRef] [PubMed]
- Molina, E.; Papadopoulou, A.; Ledward, D.A. Emulsifying properties of high pressure treated soy protein isolate and 7S and 11S globulins. Food Hydrocoll. 2001, 15, 263–269. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, J.; Hansen, E.T.; Bredie, W.L.P.; Lametsch, R. Structural characteristics of low bitter and high umami protein hydrolysates prepared from bovine muscle and porcine plasma. Food Chem. 2018, 257, 163–171. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Vasanthan, T.; Chen, L. Impacts of pH and heating temperature on formation mechanisms and properties of thermally induced canola protein gels. Food Hydrocoll. 2014, 40, 225–236. [Google Scholar] [CrossRef]
- Segat, A.; Misra, N.N.; Fabbro, A.; Buchini, F.; Lippe, G.; Cullen, P.J.; Innocente, N. Effects of ozone processing on chemical, structural and functional properties of whey protein isolate. Food Res. Int. 2014, 66, 365–372. [Google Scholar] [CrossRef]
- Cui, X.; Xiong, Y.L.; Kong, B.; Zhao, X.; Liu, N. Hydroxyl radical-stressed whey protein isolate: Chemical and structural properties. Food Bioprocess Technol. 2012, 5, 2454–2461. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Tavares, W.P.S.; Dong, S.; Yang, Y.; Zeng, M.; Zhao, Y. Influence of cooking methods on protein modification and in vitro digestibility of hairtail (Thichiurus lepturus) fillets. LWT Food Sci. Technol. 2018, 96, 476–481. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | 0.1 MPa | 100 MPa | 200 MPa | 300 MPa | 400 MPa | 500 MPa |
|---|---|---|---|---|---|---|
| 3 | 5.74 ± 0.48c | 8.12 ± 0.22b | 9.78 ± 0.11a | 10.67 ± 0.99a | 9.93 ± 0.06a | 9.92 ± 0.48a |
| 4 | 2.96 ± 0.34a | 2.67 ± 0.22a | 2.95 ± 0.23a | 3.16 ± 0.15a | 2.76 ± 0.19a | 3.38 ± 0.83a |
| 5 | 3.12 ± 0.41e | 6.32 ± 0.34d | 6.75 ± 0.11cd | 9.16 ± 0.21a | 7.44 ± 0.15bc | 7.57 ± 0.12b |
| 6 | 4.62 ± 0.79e | 10.91 ± 0.24d | 13.73 ± 0.10c | 18.32 ± 0.25b | 19.84 ± 0.14a | 19.70 ± 0.1a |
| 7 | 10.53 ± 0.96e | 12.18 ± 0.20d | 15.19 ± 0.10c | 18.85 ± 0.31b | 20.83 ± 0.27a | 19.92 ± 0.14ab |
| 8 | 12.57 ± 0.12b | 14.73 ± 0.22b | 15.60 ± 0.42b | 19.76 ± 0.16a | 21.62 ± 0.70a | 20.18 ± 0.29a |
| 9 | 17.41 ± 0.15c | 17.98 ± 0.50c | 18.52 ± 0.58c | 21.75 ± 0.27b | 24.26 ± 0.13a | 24.18 ± 0.54a |
| 10 | 23.22 ± 0.54b | 21.78 ± 0.14bc | 20.42 ± 0.29c | 23.90 ± 0.68b | 28.05 ± 0.97a | 28.92 ± 0.32a |
| Sample | α-Helix | β-Sheet | β-Turn | Random Coil |
|---|---|---|---|---|
| 0.1 MPa | 28.27 ± 0.47a | 15.68 ± 0.67a | 31.16 ± 0.16b | 24.89 ± 0.19b |
| 100 MPa | 27.15 ± 0.10ab | 12.54 ± 0.98b | 31.77 ± 0.29b | 28.54 ± 0.64a |
| 200 MPa | 27.05 ± 0.29b | 12.54 ± 0.23b | 32.05 ± 0.47a | 28.36 ± 0.67a |
| 300 MPa | 26.51 ± 0.36bc | 12.14 ± 0.01b | 33.24 ± 0.63a | 28.11 ± 0.54a |
| 400 MPa | 26.35 ± 1.01bc | 11.91 ± 0.17b | 33.33 ± 1.26a | 28.40 ± 0.33a |
| 500 MPa | 25.67 ± 0.97c | 11.90 ± 0.46b | 33.75 ± 0.47a | 28.69 ± 0.73a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Jiang, S.; Li, Y.; Xu, Y.; Zhang, T.; Zhang, F.; Feng, X.; Zhao, Y.; Zeng, M. Effects of High Pressure Modification on Conformation and Digestibility Properties of Oyster Protein. Molecules 2019, 24, 3273. https://doi.org/10.3390/molecules24183273
Wang R, Jiang S, Li Y, Xu Y, Zhang T, Zhang F, Feng X, Zhao Y, Zeng M. Effects of High Pressure Modification on Conformation and Digestibility Properties of Oyster Protein. Molecules. 2019; 24(18):3273. https://doi.org/10.3390/molecules24183273
Chicago/Turabian StyleWang, Runfang, Suisui Jiang, Yujin Li, Yunsheng Xu, Tietao Zhang, Fan Zhang, Xue Feng, Yuanhui Zhao, and Mingyong Zeng. 2019. "Effects of High Pressure Modification on Conformation and Digestibility Properties of Oyster Protein" Molecules 24, no. 18: 3273. https://doi.org/10.3390/molecules24183273
APA StyleWang, R., Jiang, S., Li, Y., Xu, Y., Zhang, T., Zhang, F., Feng, X., Zhao, Y., & Zeng, M. (2019). Effects of High Pressure Modification on Conformation and Digestibility Properties of Oyster Protein. Molecules, 24(18), 3273. https://doi.org/10.3390/molecules24183273