
Proanthocyanidin-Rich Fractions from Red Rice Extract Enhance TNF-α-Induced Cell Death and Suppress Invasion of Human Lung Adenocarcinoma Cell A549


 and
and
Abstract
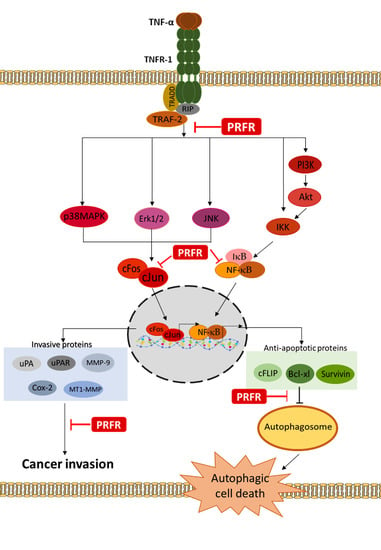
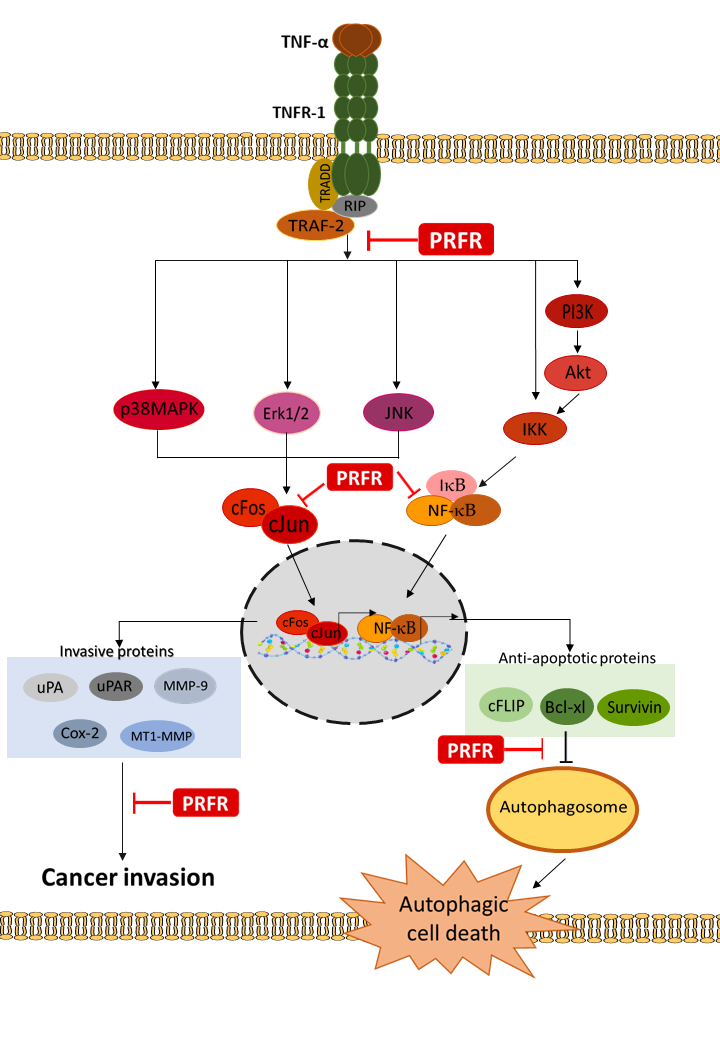
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
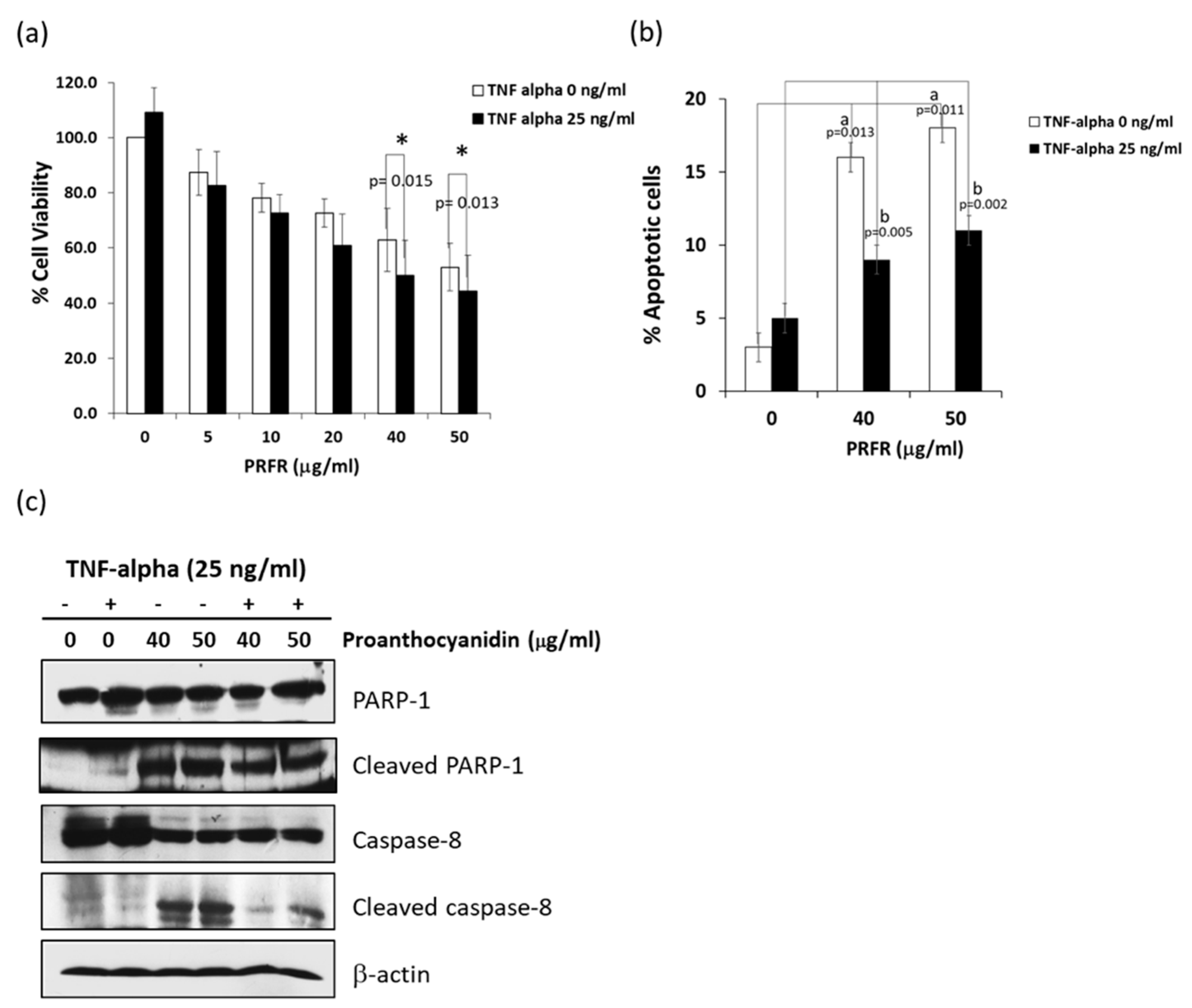
2.1. PRFR Enhanced TNF-α-Induced Cytotoxicity in A549 Lung Adenocarcinoma Cells
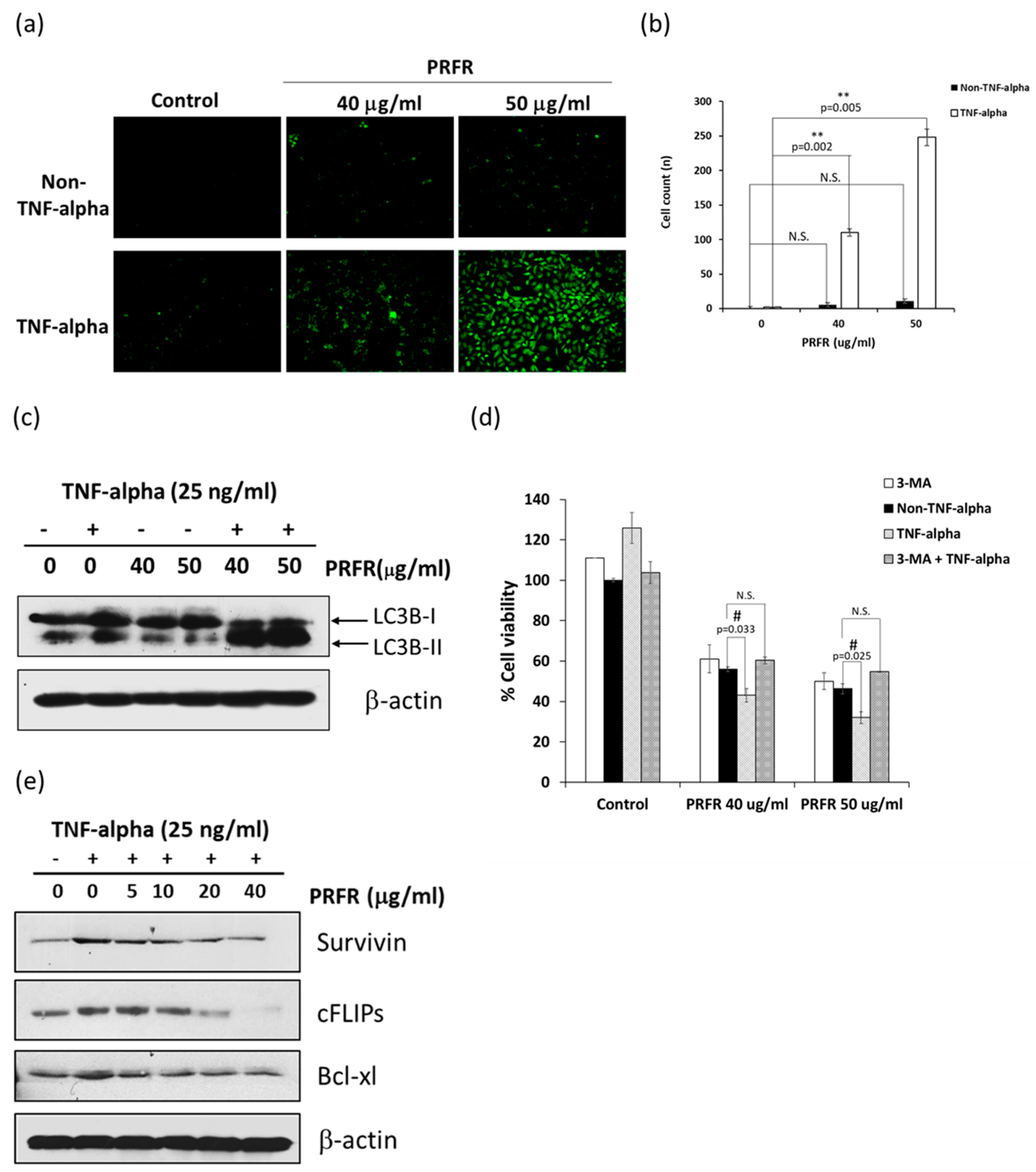
2.2. PRFR Potentiates TNF-α-Induced Autophagy
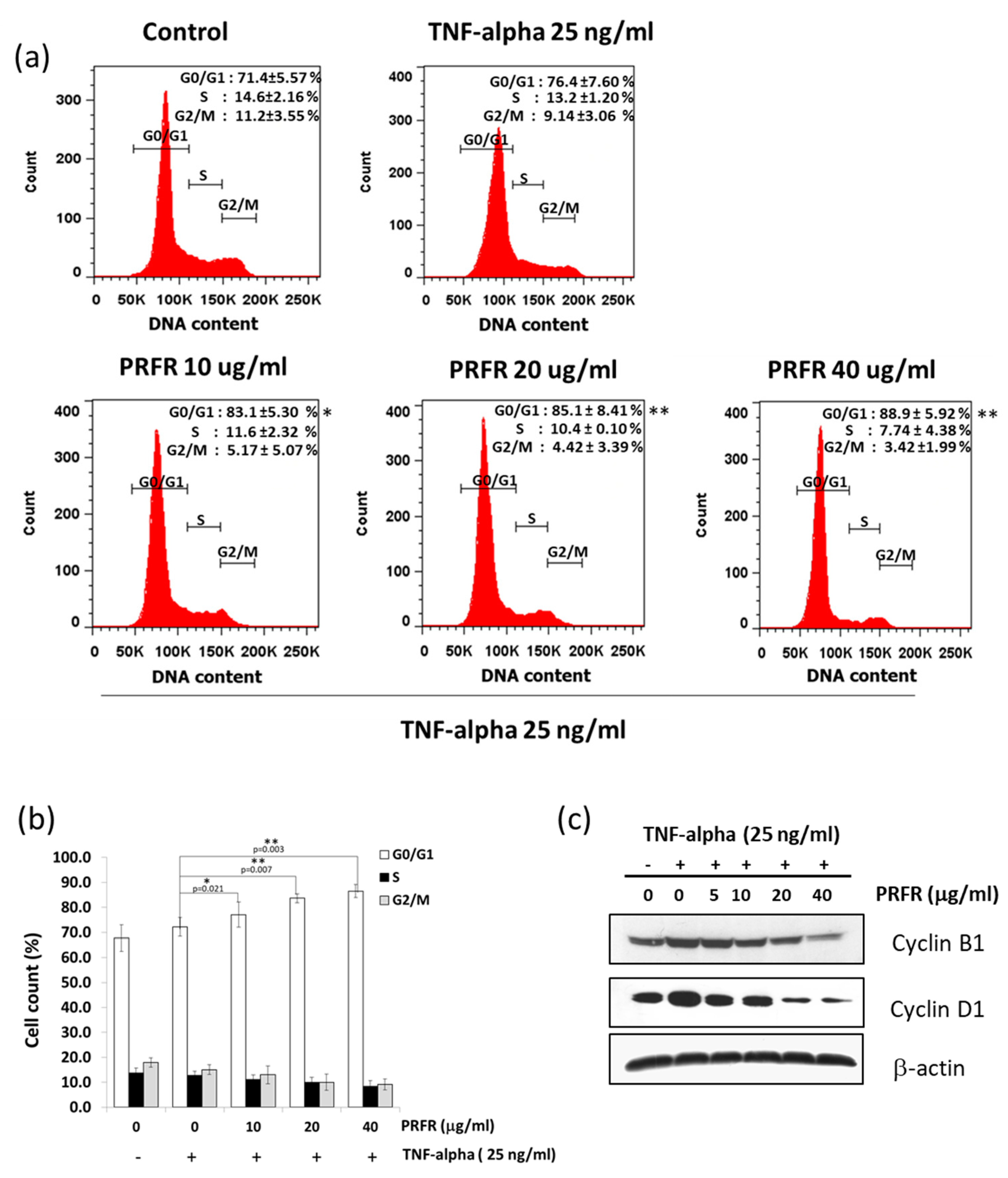
2.3. Effect of PRFRon TNF-α-Induced Cell Proliferation
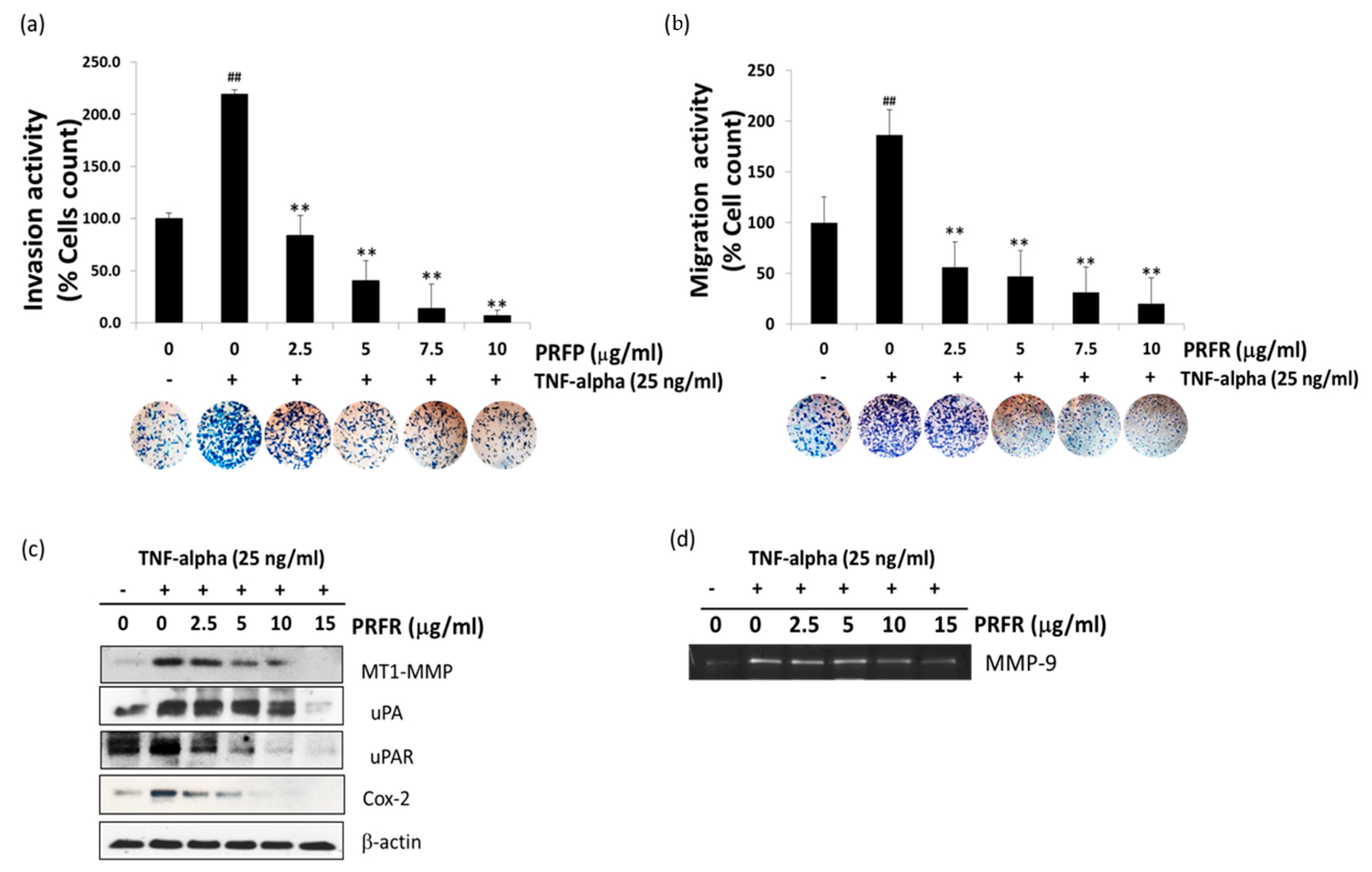
2.4. PRFR Inhibited TNF-α-Induced A549 Cell Invasion and Migration
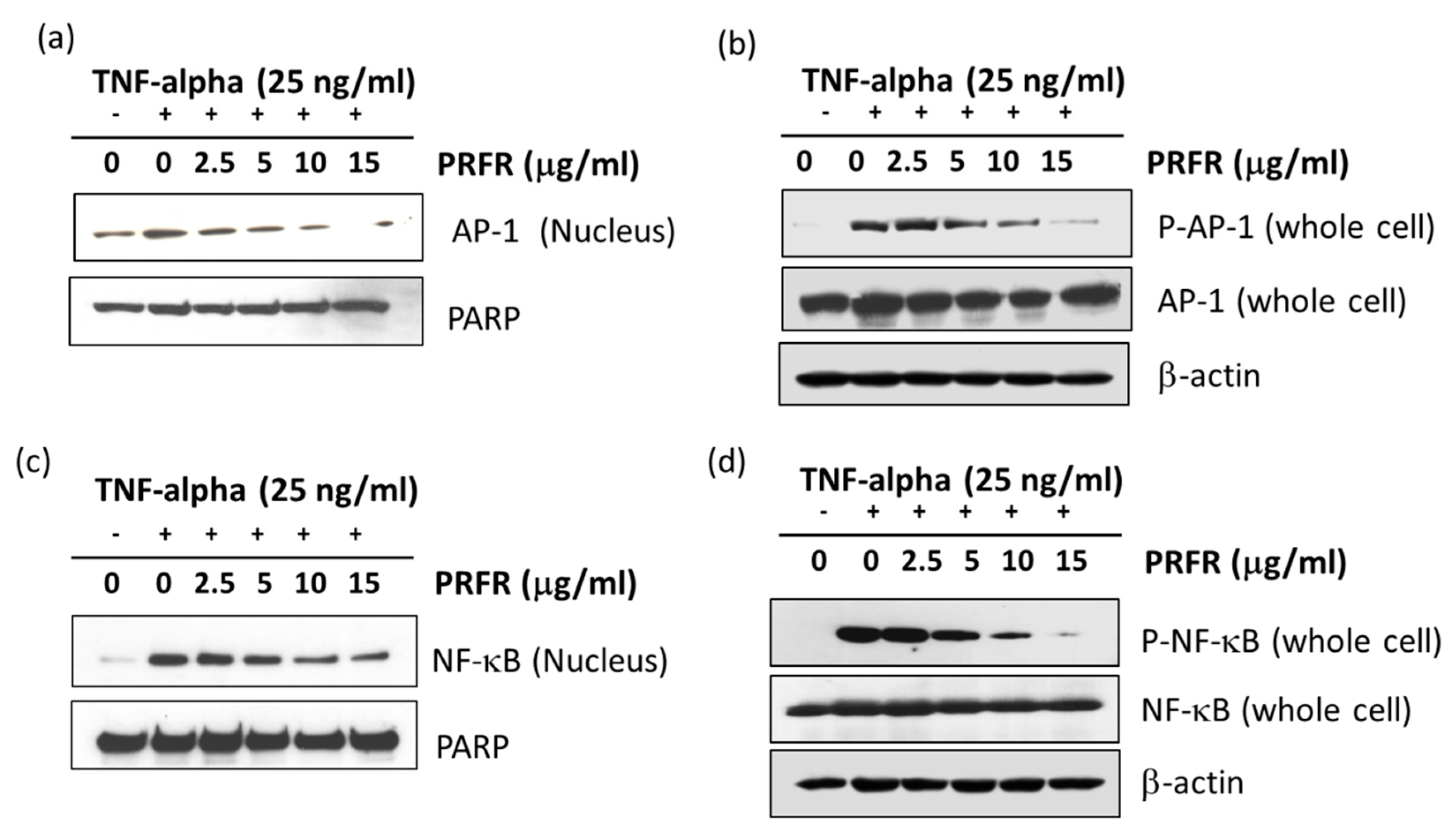
2.5. Effect of PRFR on TNF-α-Induced NF-κB and AP-1 Activation
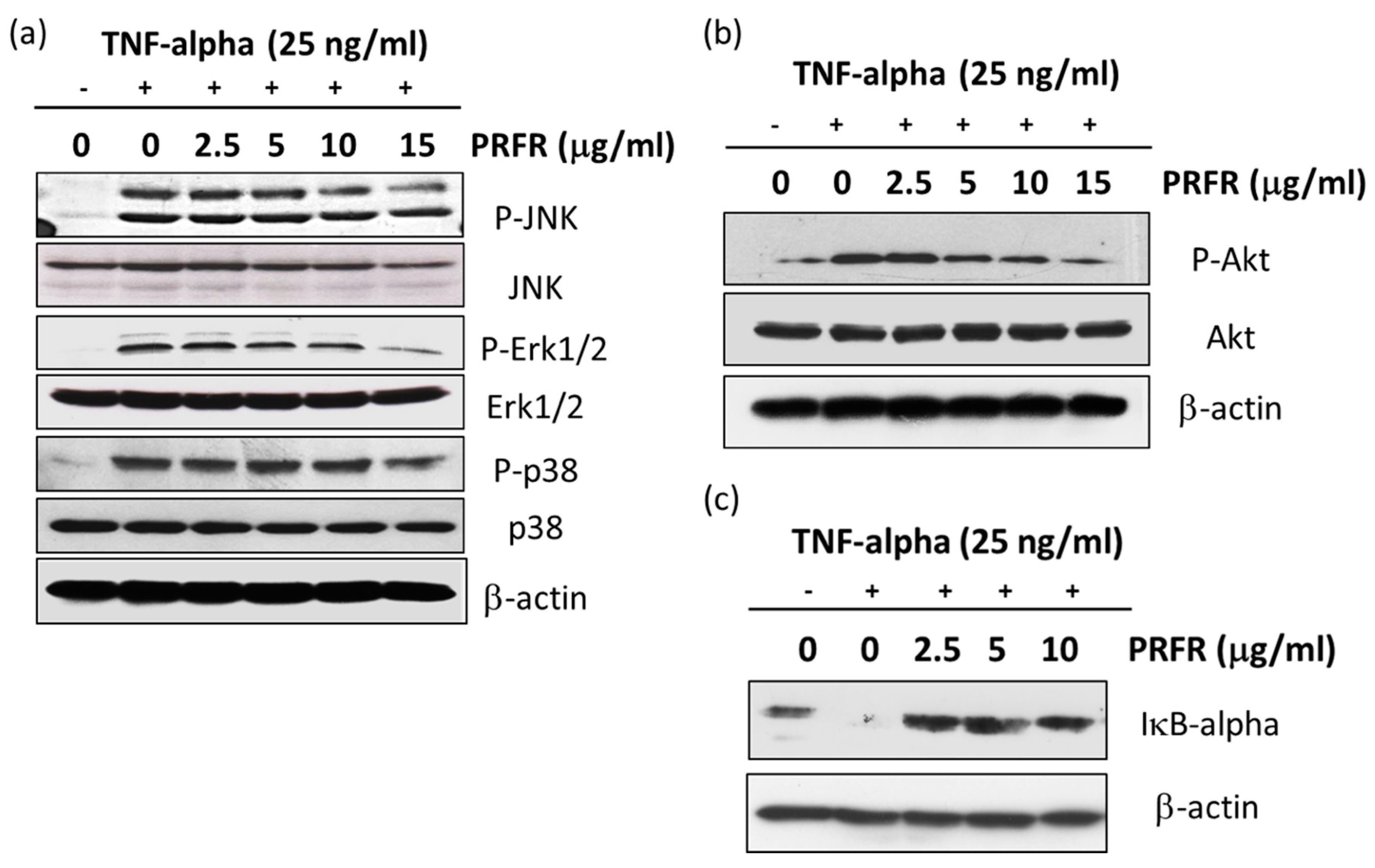
2.6. Effect of PRFR on TNF-α-Induced MAPK, Akt, and IκB-α Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of Proanthocyanidin-Rich Fraction from Red Rice Extract
4.3. Cell Cultures
4.4. Cell Viability Assay
4.5. Cell Cycle Arrest Assay
4.6. Apoptosis Assay
4.7. Extraction of Nuclear and Whole-Cell Lysate
4.8. Western Blotting Analysis
4.9. Monodansylcadaverine Staining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farbicka, P.; Nowicki, A. Palliative care in patients with lung cancer. Contemp. Oncol. (Pozn) 2013, 17, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A., III; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Beelen, R.; Hoek, G.; van den Brandt, P.A.; Goldbohm, R.A.; Fischer, P.; Schouten, L.J.; Armstrong, B.; Brunekreef, B. Long-Term Exposure to Traffic-Related Air Pollution and Lung Cancer Risk. Epidemiology 2008, 19, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-B.; Shim, J.-Y.; Park, B.; Lee, Y.-J. Long-Term Exposure to Air Pollutants and Cancer Mortality: A Meta-Analysis of Cohort Studies. Int. J. Environ. Res. Public Health 2018, 15, 2608. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A. Inflammation and Lung Cancer Oxidative Stress, ROS, and DNA Damage. React. Oxyg. Spec. Biol. Hum. Health 2016, 1, 215–223. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.S.; Liu, L.; Qin, Y.W. IL-6 and TNF-alpha promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Perez-Gracia, J.L.; Prior, C.; Guillén-Grima, F.; Segura, V.; Gonzalez, A.; Panizo, A.; Melero, I.; Grande-Pulido, E.; Gurpide, A.; Gil-Bazo, I. Identification of TNF-α and MMP-9 as potential baseline predictive serum markers of sunitinib activity in patients with renal cell carcinoma using a human cytokine array. Br. J. Cancer 2009, 101, 1876. [Google Scholar] [CrossRef] [PubMed]
- Devin, A.; Lin, Y.; Liu, Z.G. The role of the death-domain kinase RIP in tumour-necrosis-factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003, 4, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharm. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Herreweghe, F.; Festjens, N.; Declercq, W.; Vandenabeele, P. Tumor necrosis factor-mediated cell death: To break or to burst, that’s the question. Cell. Mol. Life Sci. 2010, 67, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J. Carcinog. Mutagen. 2013. [Google Scholar] [CrossRef]
- Sahni, S.; Merlot, A.M.; Krishan, S.; Jansson, P.J.; Richardson, D.R. Gene of the month: BECN1. J. Clin. Pathol. 2014, 67, 656–660. [Google Scholar] [CrossRef]
- Gabetta, B.; Fuzzati, N.; Griffini, A.; Lolla, E.; Pace, R.; Ruffilli, T.; Peterlongo, F. Characterization of proanthocyanidins from grape seeds. Fitoterapia 2000, 71, 162–175. [Google Scholar] [CrossRef]
- Limtrakul, P.; Yodkeeree, S.; Pitchakarn, P.; Punfa, W. Anti-inflammatory effects of proanthocyanidin-rich red rice extract via suppression of MAPK, AP-1 and NF-κB pathways in Raw 264.7 macrophages. Nutr. Res. Pract. 2016, 10, 251–258. [Google Scholar] [CrossRef]
- Pintha, K.; Yodkeeree, S.; Limtrakul, P. Proanthocyanidin in red rice inhibits MDA-MB-231 breast cancer cell invasion via the expression control of invasive proteins. Biol. Pharm. Bull. 2015, 38, 571–581. [Google Scholar] [CrossRef]
- Upanan, S.; Yodkeeree, S.; Thippraphan, P.; Punfa, W.; Wongpoomchai, R.; Limtrakul Dejkriengkraikul, P. The Proanthocyanidin-Rich Fraction Obtained from Red Rice Germ and Bran Extract Induces HepG2 Hepatocellular Carcinoma Cell Apoptosis. Molecules 2019, 24, 813. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Springer: Berlin/Heidelberg, Germany, 2008; pp. 77–88. [Google Scholar]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome Proteins LC3A, LC3B and LC3C Have Distinct Subcellular Distribution Kinetics and Expression in Cancer Cell Lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Gebhardt, S.; Prior, R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J. Nutr. 2004, 134, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891. [Google Scholar] [CrossRef] [PubMed]
- Sivaprasad, U.; Basu, A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-α-induced cell death in MCF-7 cells. J. Cell. Mol. Med. 2008, 12, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- FBauvy, D.-M.M.M.J. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar]
- Zhu, J.; Cai, Y.; Xu, K.; Ren, X.; Sun, J.; Lu, S.; Chen, J.; Xu, P. Beclin1 overexpression suppresses tumor cell proliferation and survival via an autophagy-dependent pathway in human synovial sarcoma cells. Oncol. Rep. 2018, 40, 1927–1936. [Google Scholar] [CrossRef]
- Radeff-Huang, J.; Seasholtz, T.M.; Chang, J.W.; Smith, J.M.; Walsh, C.T.; Brown, J.H. Tumor necrosis factor-α-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression. J. Biol. Chem. 2007, 282, 863–870. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A role in cancer stem cell survival and repopulation of cancer cells during therapy. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016, 291, 5068–5079. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Codogno, P. Regulation of autophagy by NF-kappaB transcription factor and reactives oxygen species. Autophagy 2007, 3, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Souquère, S.; Pierron, G.; Codogno, P. NF-κB activation represses tumor necrosis factor-α-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-Y.; Lee, J.-K.; Jeon, Y.-K.; Kim, C.-W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Pratheeshkumar, P.; Sreekala, C.; Zhang, Z.; Budhraja, A.; Ding, S.; Son, Y.-O.; Wang, X.; Hitron, A.; Hyun-Jung, K.; Wang, L. Cancer prevention with promising natural products: Mechanisms of action and molecular targets. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. Anti-Cancer Agents) 2012, 12, 1159–1184. [Google Scholar] [CrossRef]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim. Biophys. Acta (Bba) Rev. Cancer 2014, 1846, 342–352. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subkamkaew, C.; Limtrakul, P.; Yodkeeree, S. Proanthocyanidin-Rich Fractions from Red Rice Extract Enhance TNF-α-Induced Cell Death and Suppress Invasion of Human Lung Adenocarcinoma Cell A549. Molecules 2019, 24, 3393. https://doi.org/10.3390/molecules24183393
Subkamkaew C, Limtrakul P, Yodkeeree S. Proanthocyanidin-Rich Fractions from Red Rice Extract Enhance TNF-α-Induced Cell Death and Suppress Invasion of Human Lung Adenocarcinoma Cell A549. Molecules. 2019; 24(18):3393. https://doi.org/10.3390/molecules24183393
Chicago/Turabian StyleSubkamkaew, Chayaporn, Pornngarm Limtrakul (Dejkriengkraikul), and Supachai Yodkeeree. 2019. "Proanthocyanidin-Rich Fractions from Red Rice Extract Enhance TNF-α-Induced Cell Death and Suppress Invasion of Human Lung Adenocarcinoma Cell A549" Molecules 24, no. 18: 3393. https://doi.org/10.3390/molecules24183393
APA StyleSubkamkaew, C., Limtrakul, P., & Yodkeeree, S. (2019). Proanthocyanidin-Rich Fractions from Red Rice Extract Enhance TNF-α-Induced Cell Death and Suppress Invasion of Human Lung Adenocarcinoma Cell A549. Molecules, 24(18), 3393. https://doi.org/10.3390/molecules24183393