
A Complete Study of Farrerol Metabolites Produced In Vivo and In Vitro

 ,
,
Abstract
:1. Introduction
2. Results
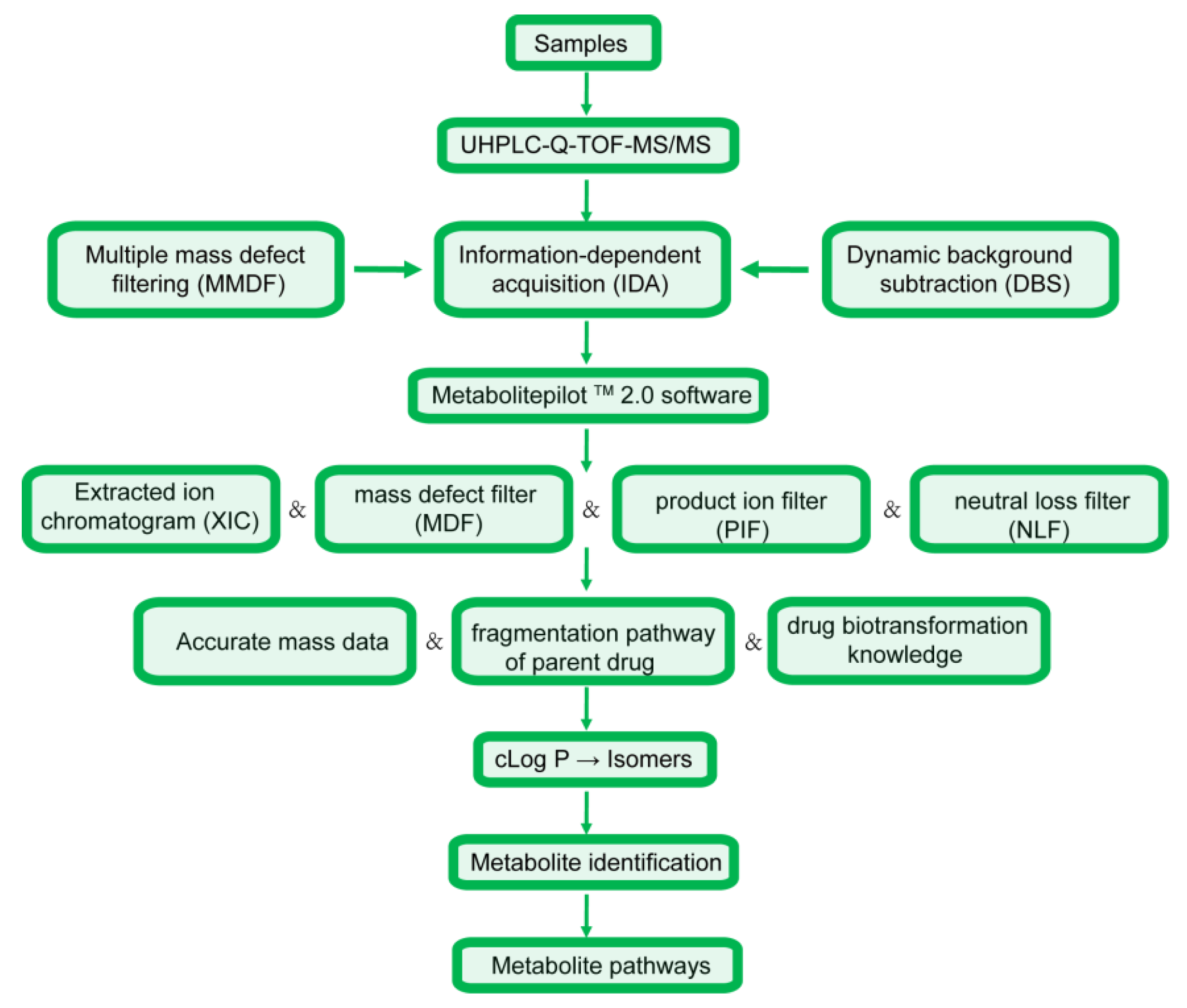
2.1. Analytical Procedure
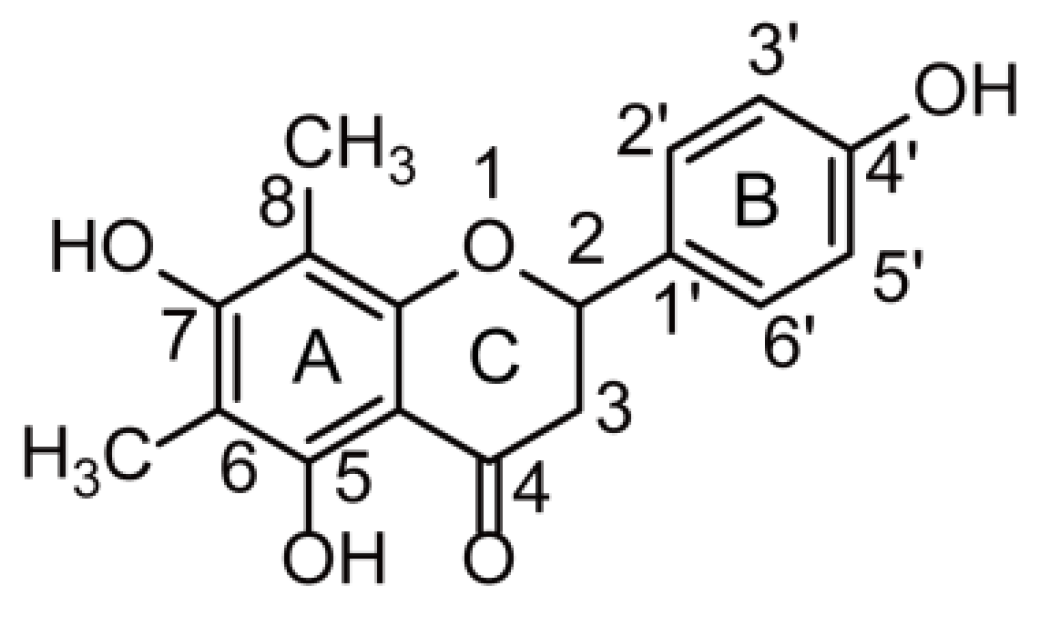
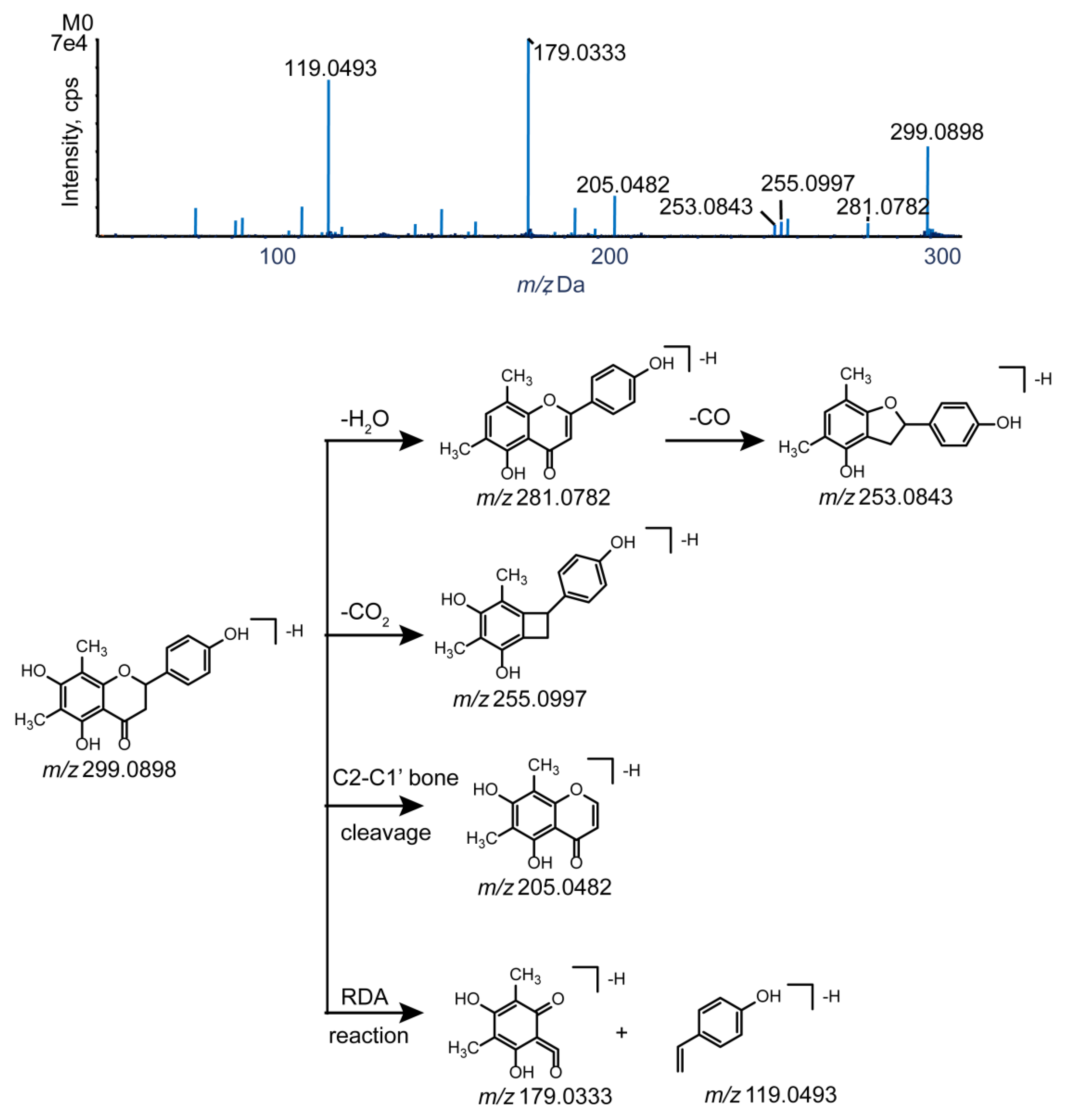
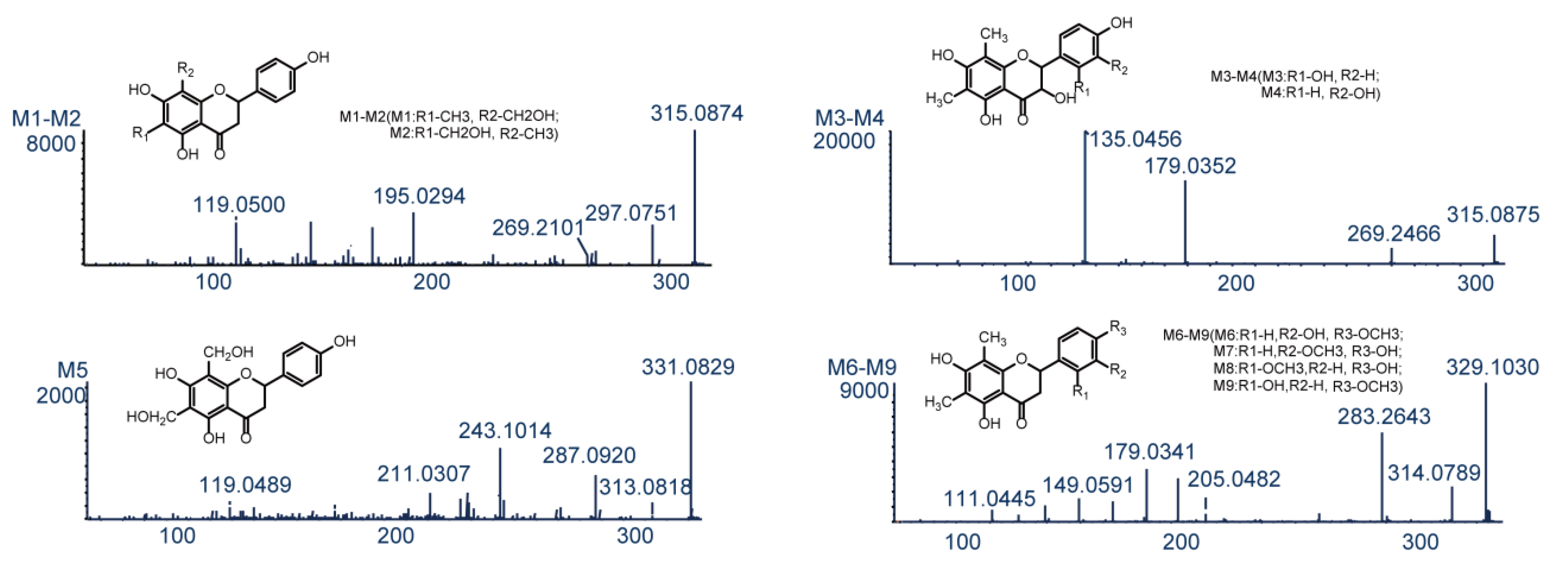
2.2. Farrerol Fragmentation Patterns
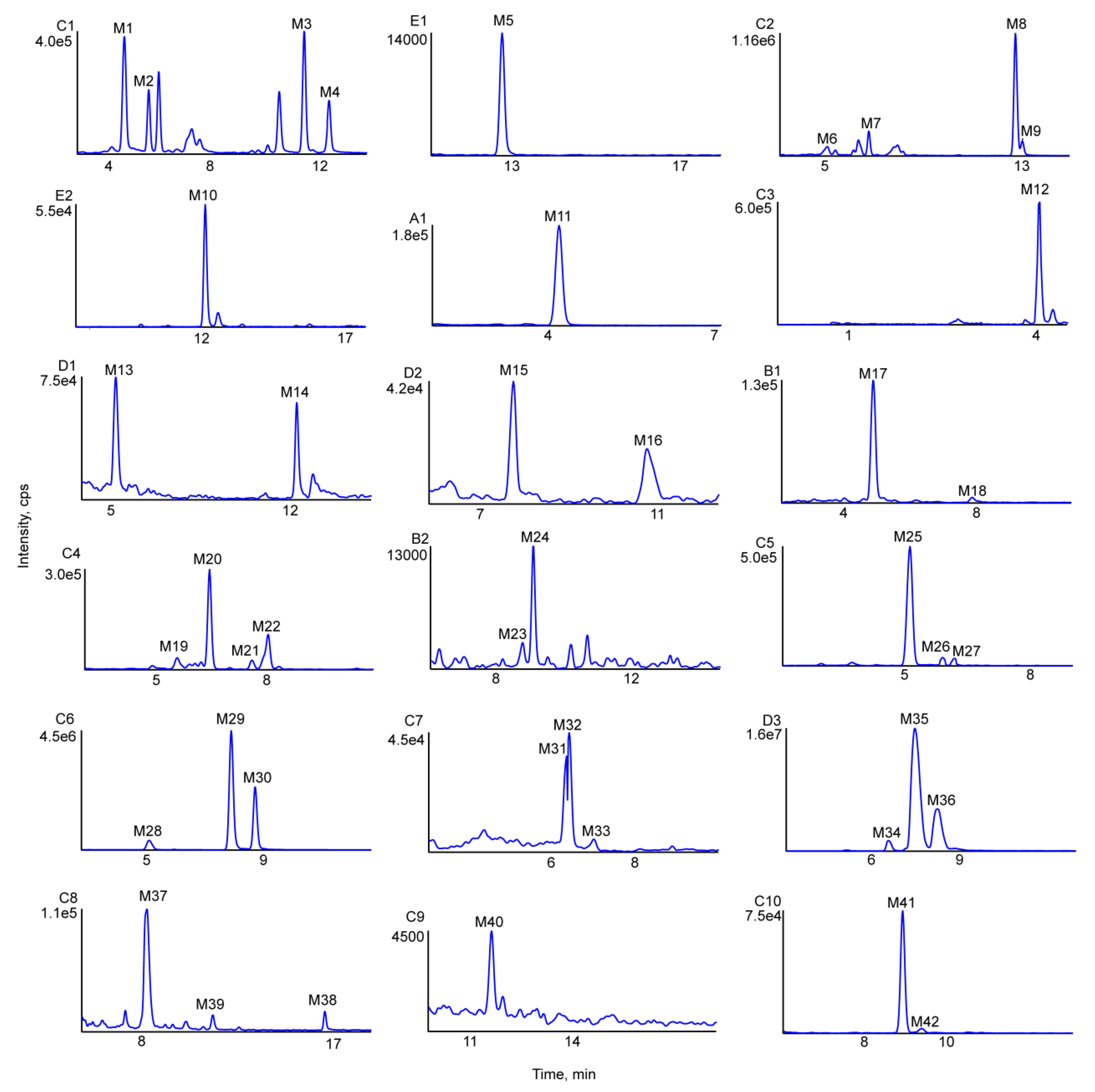
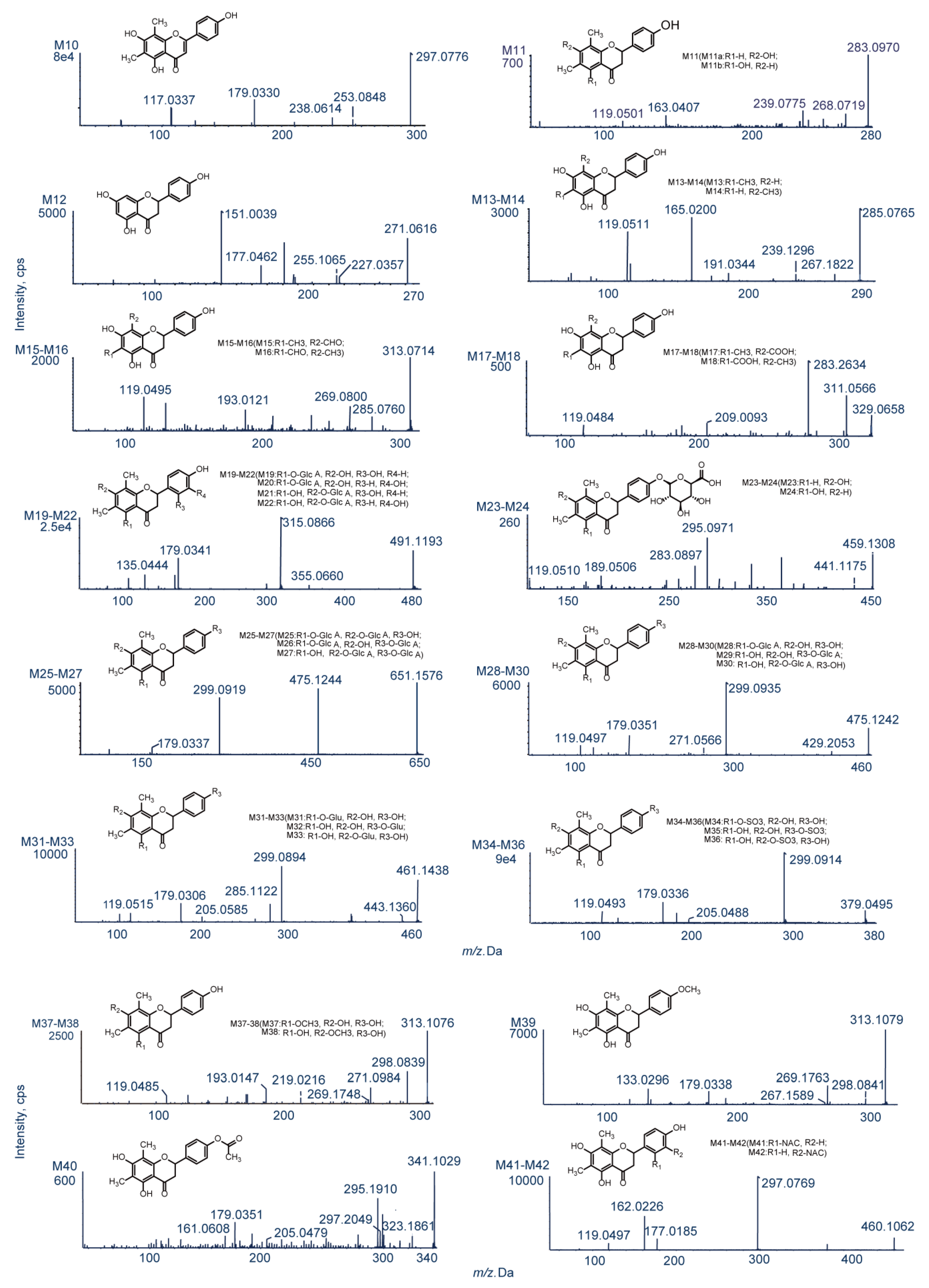
2.3. Identification of Phase I Metabolites
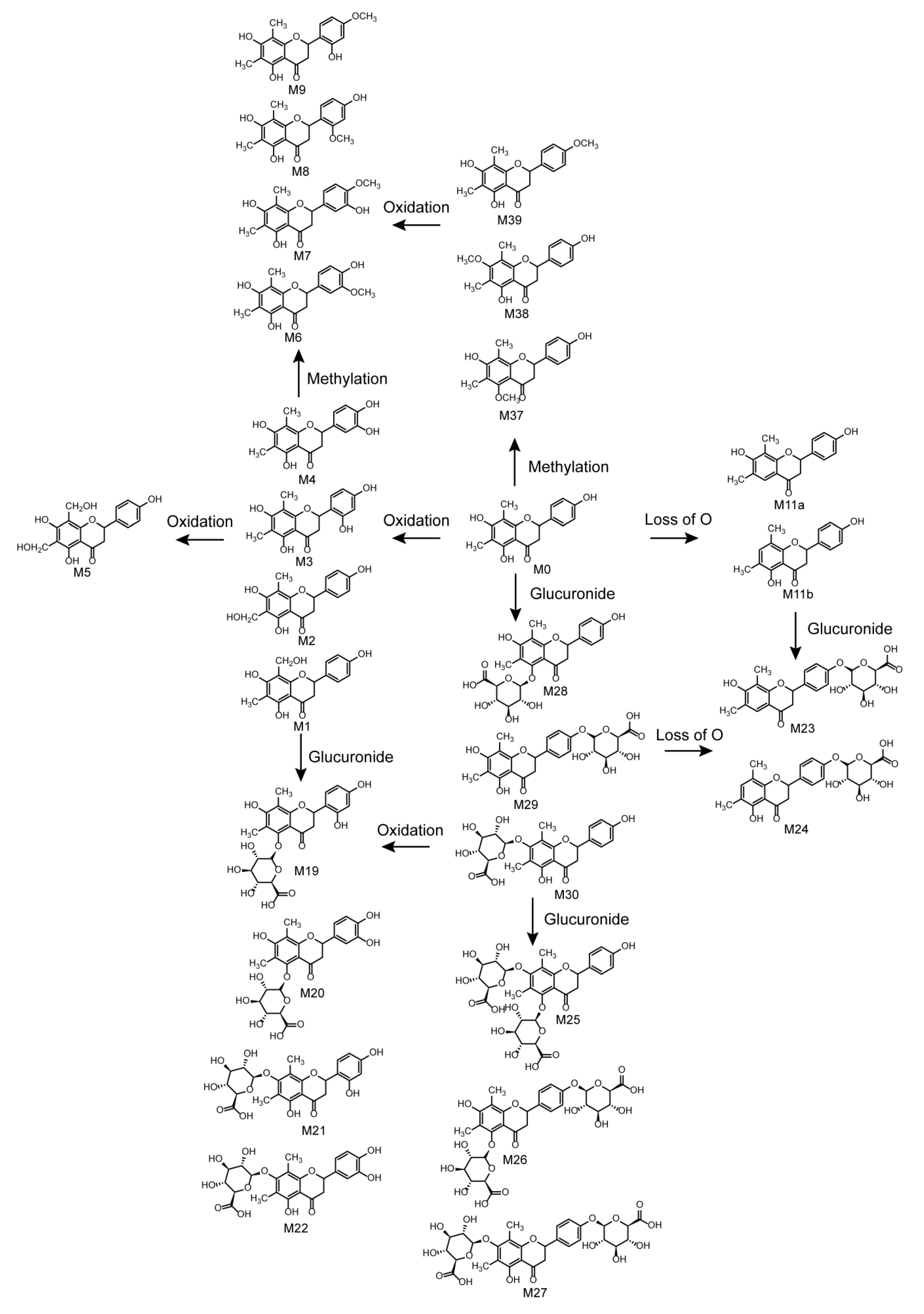
2.3.1. Oxidation Reactions
2.3.2. Loss of O (deoxygenation)
2.3.3. Demethylation
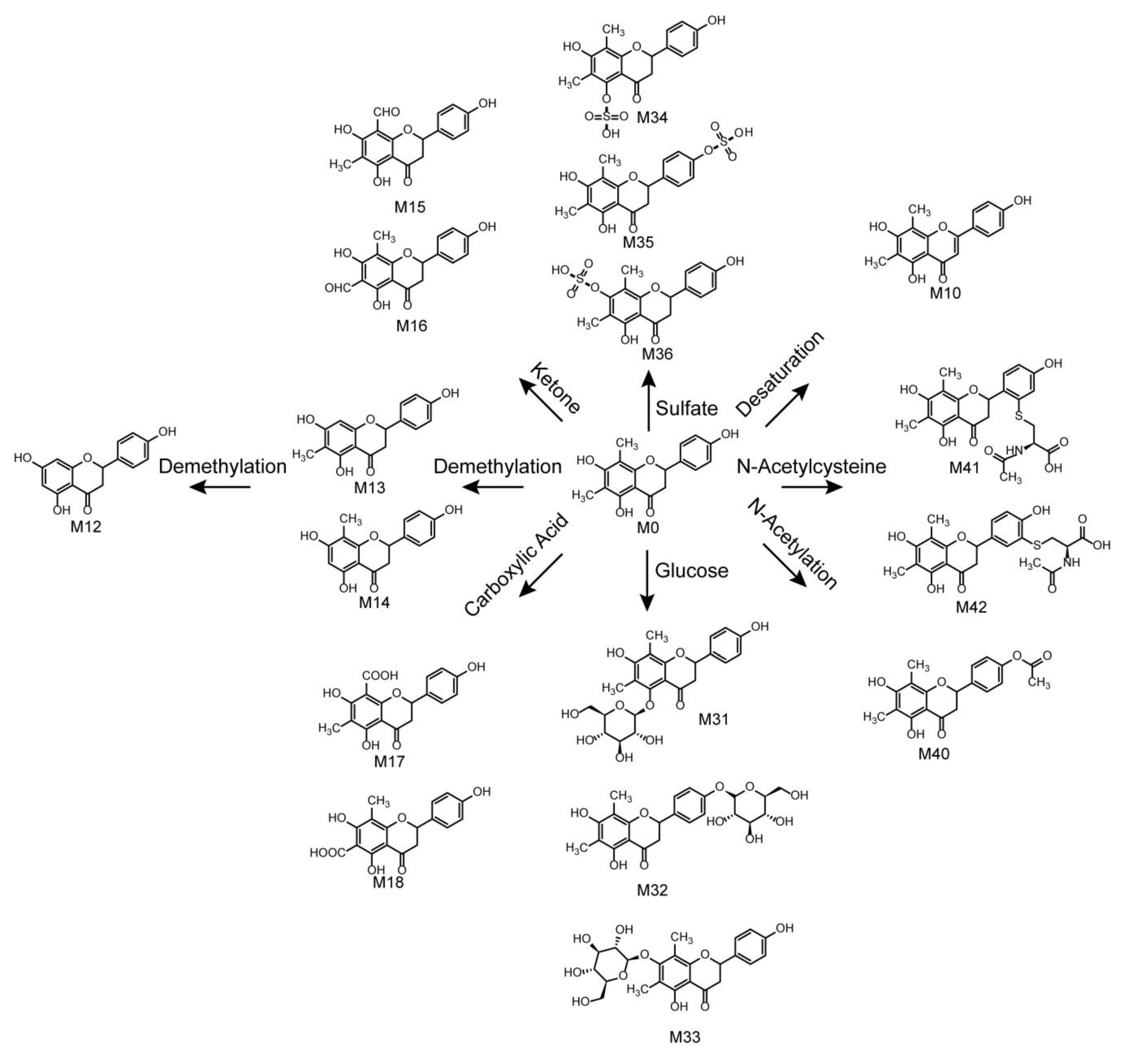
2.4. Identification of Phase II Metabolites
2.5. Metabolic Pathways of Farrerol
3. Discussion
3.1. Optimizations of Liquid Conditions and Mass Conditions
3.2. Hypotheses of Possible Enzymes
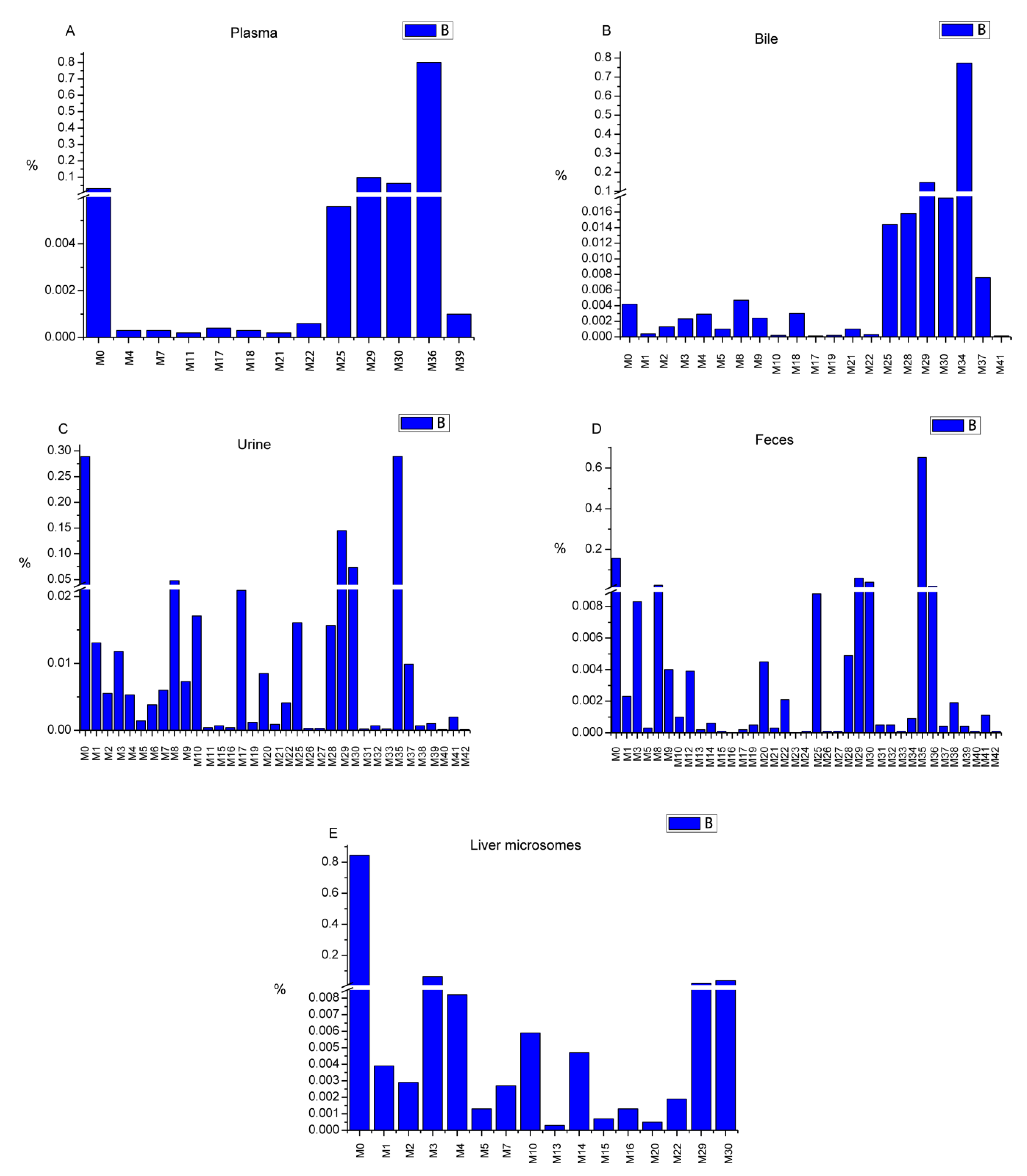
3.3. Semi-quantitative Analysis
3.4. Metabolite Analysis
3.4.1. Comparison of In Vivo and In Vitro Metabolism
3.4.2. Reference to Previous Studies
3.4.3. Metabolic Activities of Farrerol
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Instruments and Conditions
4.3. Animals and Drug Administration
4.4. In Vivo Sample Collection and Pretreatment
4.5. In Vitro Sample Incubation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Justesen, U.; Knuthsen, P.; Leth, T. Quantitative analysis of flavonols, flavones, and flavanones in fruits, vegetables and beverages by high-performance liquid chromatography with photo-diode array and mass spectrometric detection. J. Chromatogr. A 1998, 799, 101–110. [Google Scholar] [CrossRef]
- Cao, Y.; Lou, C.; Fang, Y.; Ye, J. Determination of active ingredients of Rhododendron dauricum L. by capillary electrophoresis with electrochemical detection. J. Chromatogr. A 2002, 943, 153–157. [Google Scholar] [CrossRef]
- Hostetler, G.L.; Ralston, R.A.; Schwartz, S.J. Flavones: Food Sources, Bioavailability, Metabolism, and Bioactivity. Adv. Nutr. 2017, 8, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.K.; Pratap, R. The biological potential of flavones. Nat. Prod. Rep. 2010, 27, 1571–1593. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M. A semi-empirical study of flavone compounds with antioxidant efficiency. Afr. J. Pharm. Pharmacol. 2011, 5, 2140–2144. [Google Scholar] [CrossRef] [Green Version]
- Hostetler, G.; Riedl, K.; Cardenas, H.; Diosa-Toro, M.; Arango, D.; Schwartz, S.; Doseff, A.I. Flavone deglycosylation increases their anti-inflammatory activity and absorption. Mol. Nutr. Food Res. 2012, 56, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Jiang, W.B.; Xie, M.X. Flavonoids: Recent Advances as Anticancer Drugs. Recent Pat. Anti-Cancer Drug Discov. 2010, 5, 152–164. [Google Scholar] [CrossRef]
- Cao, Y.; Chu, Q.; Ye, J. Chromatographic and electrophoretic methods for pharmaceutically active compounds in Rhododendron dauricum. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 812, 231–240. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, J.; Zhuang, Y.; Han, G. Anti-inflammatory effects of farrerol on IL-1beta-stimulated human osteoarthritis chondrocytes. Eur. J. Pharmacol. 2015, 764, 443–447. [Google Scholar] [CrossRef]
- Qiu, J.; Xiang, H.; Hu, C.; Wang, Q.; Dong, J.; Li, H.; Luo, M.; Wang, J.; Deng, X. Subinhibitory concentrations of farrerol reduce alpha-toxin expression in Staphylococcus aureus. FEMS Microbiol. Lett. 2011, 315, 129–133. [Google Scholar] [CrossRef]
- Ci, X.; Lv, H.; Wang, L.; Wang, X.; Peng, L.; Qin, F.X.; Cheng, G. The antioxidative potential of farrerol occurs via the activation of Nrf2 mediated HO-1 signaling in RAW 264.7 cells. Chem. Biol. Interact. 2015, 239, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, S.; Lu, J.; Sun, S.; Song, B.; Xu, L.; Yang, Z.; Guan, S. Investigation of effects of farrerol on suppression of murine T lymphocyte activation in vitro and in vivo. Int. Immunopharmacol. 2013, 16, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hou, X.; Zhang, M.; Liang, T.; Zhi, J.; Han, L.; Li, Q. Relaxation of rat aorta by farrerol correlates with potency to reduce intracellular calcium of VSMCs. Int. J. Mol. Sci. 2014, 15, 6641–6656. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Li, Y.; Chen, G.; Fu, S.; He, D.; Huang, B.; Wei, L.; Lin, Y.; Guo, Y.; Hu, G. Farrerol Ameliorates TNBS-Induced Colonic Inflammation by Inhibiting ERK1/2, JNK1/2, and NF-kappaB Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 2037. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, P.; Wang, J.H.; Li, L.; Gong, J.L.; Yao, H. Farrerol overcomes the invasiveness of lung squamous cell carcinoma cells by regulating the expression of inducers of epithelial mesenchymal transition. Microb. Pathog. 2019, 131, 277. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Liang, T.; Wang, X.; Ban, S.; Han, L.; Li, Q. Apoptosis induced by farrerol in human gastric cancer SGC-7901 cells through the mitochondrial-mediated pathway. Eur. J. Cancer Prev. 2015, 24, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hou, X.; Zhang, K.; Li, Q. Farrerol Modulates Aorta Gene Expression Profile in Spontaneously Hypertensive Rats. Planta Med. 2018, 84, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Garg, A.; Takwani, H.; Singh, S. Metabolite identification by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2011, 30, 360–387. [Google Scholar] [CrossRef]
- Tiwari, S.S.; Chavan, B.B.; Kushwah, B.S.; Yerra, N.V.; Mukesh, S.; Sangamwar, A.T.; Thaota, J.R.; Talluri, M. In vitroand in vivo investigation of metabolic fate of riociguat by HPLC-Q-TOF/MS/MS and in silico evaluation of the metabolites by ADMET predictor. J. Pharm. Biomed. Anal. 2019, 164, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, D. Metabolism of farrerol (author’s transl). Yao Xue Xue Bao (Acta Pharm Sinica) 1979, 14, 149–155. [Google Scholar]
- Feng, X.; Li, Y.; Guang, C.; Qiao, M.; Wang, T.; Chai, L.; Qiu, F. Characterization of the In Vivo and In Vitro Metabolites of Linarin in Rat Biosamples and Intestinal Flora Using Ultra-High Performance Liquid Chromatography Coupled with Quadrupole Time-of-Flight Tandem Mass Spectrometry. Molecules 2018, 23, 2140. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qi, P.; Zhou, N.; Zhao, M.; Ding, W.; Li, S.; Liu, M.; Wang, Q.; Jin, S. A pre-classification strategy based on UPLC-Triple-TOF/MS for metabolic screening and identification of Radix glehniae in rats. Anal. Bioanal. Chem. 2016, 408, 7423–7436. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ma, Y.; Liang, C.; Gao, J.; Wang, H.; Zhang, L. A Systematic Study of the Metabolites of Dietary Acacetin in Vivo and in Vitro Based on UHPLC-Q-TOF-MS/MS Analysis. J. Agric. Food Chem. 2019, 67, 5530–5543. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shang, Z.; Li, Q.; Huang, M.; He, W.; Wang, Z.; Zhang, J. Rapid Screening and Identification of Daidzein Metabolites in Rats Based on UHPLC-LTQ-Orbitrap Mass Spectrometry Coupled with Data-Mining Technologies. Molecules 2018, 23, 151. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Jin, Y.; Ma, Y.; Xie, W.; Xu, H.; Zhang, K.; Zhang, L.; Du, Y. Identification of metabolites of oridonin in rats with a single run on UPLC-Triple-TOF-MS/MS system based on multiple mass defect filter data acquisition and multiple data processing techniques. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 1006, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, W.; Wang, C.; Wang, Z.; Wang, Z.; Zhang, J. A Comprehensive Screening and Identification of Genistin Metabolites in Rats Based on Multiple Metabolite Templates Combined with UHPLC-HRMS Analysis. Molecules 2018, 23, 1862. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liang, C.; Diao, X.; Cheng, X.; Liao, M.; Zhang, L. Metabolism studies on hydroxygenkwanin and genkwanin in human liver microsomes by UHPLC-Q-TOF-MS. Xenobiotica 2018, 48, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, L.; Guo, S.; Wang, Z.; Jiang, L.; Wang, F.; Zhang, J.; Liu, B. Profiling and comparison of the metabolites of diosmetin and diosmin in rat urine, plasma and feces using UHPLC-LTQ-Orbitrap MS(n). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1124, 58–71. [Google Scholar] [CrossRef]
- Wu, M.-J.; Wu, X.-L.; Zhang, D.-Q.; Qiu, F.; Ding, L.-Q.; Ma, H.-L.; Chen, X.-Z. Metabolic profiling of quercetin in rats using ultra-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, C.; Yin, J.; Sun, Y.; Zhang, L. Identification of metabolites of liquiritin in rats by UHPLC-Q-TOF-MS/MS: Metabolic profiling and pathway comparison in vitro and in vivo. RSC Adv. 2018, 8, 11813–11827. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, J.; Liang, C.; Sun, Y.; Zhang, L. UHPLC-Q-TOF-MS/MS Method Based on Four-Step Strategy for Metabolism Study of Fisetin in Vitro and in Vivo. J. Agric. Food Chem. 2017, 65, 10959–10972. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, H.; Murayama, N.; Kakimoto, K.; Takenaka, S.; Katahira, J.; Lim, Y.R.; Kim, V.; Kim, D.; Yamazaki, H.; Komori, M.; et al. Site-specific oxidation of flavanone and flavone by cytochrome P450 2A6 in human liver microsomes. Xenobiotica 2019, 49, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Zang, M.; Gu, Y.; Liu, B. Development and validation of a sensitive UHPLC-MS/MS method for quantitative analysis of farrerol in rat plasma: Application to pharmacokinetic and bioavailability studies. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-N.; Hou, Y.-L.; Liu, B.-J.; Liu, Q.-M.; Qiao, G.-F. The Rhododendron dauricum L. Flavonoids Exert Vasodilation and Myocardial Preservation. Iran. J. Pharm. Res. 2010, 9, 303–311. [Google Scholar] [PubMed]
- Zhang, S.; Li, D.D.; Zeng, F.; Zhu, Z.H.; Song, P.; Zhao, M.; Duan, J.A. Efficient biosynthesis, analysis, solubility and anti-bacterial activities of succinylglycosylated naringenin. Nat. Prod. Res. 2019, 33, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Jin, L.; Zhang, F.; Zhang, C.; Liang, W. Naringenin as a potential immunomodulator in therapeutics. Pharmacol. Res. 2018, 135, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C.; Telford, D.E.; Edwards, J.Y.; Sutherland, B.G.; Sawyez, C.G.; Huff, M.W. Naringenin Supplementation to a Chow Diet Enhances Energy Expenditure and Fatty Acid Oxidation, and Reduces Adiposity in Lean, Pair-Fed Ldlr(-/-) Mice. Mol. Nutr. Food Res. 2019, 63, e1800833. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Chang, Y.M.; Wang, K.Y.; Chen, P.N.; Hseu, Y.C.; Chen, K.M.; Yeh, K.T.; Chen, C.J.; Hsu, L.S. Naringenin inhibited migration and invasion of glioblastoma cells through multiple mechanisms. Environ. Toxicol. 2019, 34, 233–239. [Google Scholar] [CrossRef]
- Yuan, L.; Jia, P.; Sun, Y.; Zhao, C.; Zhi, X.; Sheng, N.; Zhang, L. Study of in vitro metabolism of m-nisoldipine in human liver microsomes and recombinant cytochrome P450 enzymes by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2014, 97, 65–71. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite ID | Reaction | Molecular Formula | m/z | Error(ppm) | tR(min) | MS/MS Fragments | Clog P | Plasma | Bile | Urine | Feces | RLM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1 | Oxidation | C17H16O6 | 315.0874 | 0 | 4.75 | 297.0751, 269.2101, 195.0294, 119.0500 | 1.75585 | -b | + | +a | + | + |
| M2 | Oxidation | C17H16O6 | 315.0874 | 0 | 5.69 | 297.0748, 269.0793, 195.0283, 119.0496 | 1.75585 | - | + | + | - | + |
| M3 | Oxidation | C17H16O6 | 315.0875 | 0.3 | 11.65 | 269.2466, 179.0352, 135.0456 | 2.57585 | - | + | + | + | + |
| M4 | Oxidation | C17H16O6 | 315.0876 | 0.6 | 12.62 | 269.2126, 179.0354, 135.0455 | 2.69585 | + | + | + | - | + |
| M5 | Dioxidation | C17H16O7 | 331.0829 | 1.8 | 12.84 | 313.0818, 287.0920, 211.0307, 119.0489 | 0.218852 | - | + | + | + | + |
| M6 | Oxidation and Methylation | C18H18O6 | 329.1030 | −0.1 | 5.04 | 314.0789, 283.2643, 205.0482, 179.0341, 149.0591 | 3.14205 | - | + | + | - | - |
| M7 | Oxidation and Methylation | C18H18O6 | 329.1029 | −0.6 | 6.70 | 314.0769, 283.2606, 205.0480, 179.0331, 149.0587 | 3.14205 | + | + | + | - | + |
| M8 | Oxidation and Methylation | C18H18O6 | 329.1040 | 2.9 | 12.78 | 314.0762, 283.2602, 205.0476, 179.0328, 149.0584 | 3.34205 | - | - | + | + | - |
| M9 | Oxidation and Methylation | C18H18O6 | 329.1031 | 0.1 | 13.06 | 314.0769, 283.2611, 205.0480, 179.0331, 149.0587 | 3.39205 | - | - | + | + | - |
| M10 | Desaturation | C17H14O5 | 297.0776 | 2.6 | 12.20 | 253.0848, 238.0614, 179.0330, 117.0337 | 3.75329 | - | + | + | + | + |
| M11ac | Loss of O | C17H16O4 | 283.0970 | −2 | 4.20 | 268.0719, 239.0775,163.0407, 119.0501, | 3.43194 | + | - | - | - | - |
| M11bc | 3.94194 | |||||||||||
| M12 | Bisdemethylation | C15H12O5 | 271.0616 | 1.6 | 4.46 | 227.0357, 225.1065, 177.0462, 151.0039 | 2.44485 | - | - | + | + | - |
| M13 | Demethylation | C16H14O5 | 285.0765 | −1.1 | 5.38 | 267.1822, 239.1296, 191.0344, 165.0200, 119.0511 | 2.84385 | - | - | - | + | + |
| M14 | Demethylation | C16H14O5 | 285.0768 | −0.1 | 12.24 | 267.1962, 239.1272, 191.0365, 165.0189, 119.0496 | 2.89385 | - | - | - | + | + |
| M15 | Ketone Formation | C17H14O6 | 313.0714 | 0.4 | 7.87 | 285.0760, 269.0800, 193.0121, 119.0495 | 2.50612 | - | - | + | + | + |
| M16 | Ketone Formation | C17H14O6 | 313.0723 | −1.3 | 10.85 | 285.0761, 269.0811, 193.0495, 119.0480 | 2.92612 | - | - | + | + | + |
| M17 | Demethylation to carboxylic acid | C17H14O7 | 329.0658 | −2.7 | 4.89 | 311.0566, 283.2634, 209.0093, 119.0484 | 2.86826 | + | + | + | + | - |
| M18 | Demethylation to carboxylic acid | C17H14O7 | 329.0662 | −1.4 | 7.90 | 311.0546, 283.2613, 209.0385, 119.0473 | 3.54826 | + | + | - | - | - |
| M19 | Oxidation and glucuronide conjugation | C23H24O12 | 491.1193 | −0.4 | 5.57 | 355.0660, 315.0866, 179.0341, 135.0444 | 0.127322 | - | + | + | + | - |
| M20 | Oxidation and glucuronide conjugation | C23H24O12 | 491.1192 | −0.6 | 6.46 | 355.0870, 315.0874, 179.0331, 135.0110 | 0.247322 | - | - | + | + | + |
| M21 | Oxidation and glucuronide conjugation | C23H24O12 | 491.1195 | −0.1 | 7.62 | 355.0872, 315.0870, 179.0335, 135.0439 | 0.637322 | + | + | + | + | - |
| M22 | Oxidation and Glucuronide Conjugation | C23H24O12 | 491.1194 | −0.2 | 8.09 | 355.0867, 315.0874, 179.0343, 135.0448 | 0.757322 | + | + | + | + | + |
| M23 | Loss of O+glucuronidation | C23H24O10 | 459.1308 | 2.5 | 8.85 | 441.1175, 295.0971, 283.0897, 189.0565, 119.0510 | 1.45664 | - | - | - | + | - |
| M24 | Loss of O+glucuronidation | C23H24O10 | 459.1286 | −2.3 | 9.03 | 441.1155, 295.0820, 283.0891, 189.0555, 119.0522 | 1.96664 | - | - | - | + | - |
| M25 | Bisglucuronide conjugation | C29H32O17 | 651.1576 | 1.4 | 5.24 | 475.1244, 299.0919, 179.0337 | −1.33398 | + | + | + | + | - |
| M26 | Bisglucuronide conjugation | C29H32O17 | 651.1578 | 1.8 | 5.85 | 475.1240, 299.0913, 179.0331 | −1.13098 | - | - | + | + | - |
| M27 | Bisglucuronide conjugation | C29H32O17 | 651.1588 | 3.3 | 6.13 | 475.1240, 299.0910, 179.0325 | −0.62098 | - | - | + | + | - |
| M28 | Glucuronidation conjugation | C23H24O11 | 475.1242 | −0.7 | 5.09 | 429.2053, 299.0935, 271.0566, 179.0351, 119.0497 | 0.844322 | - | + | + | + | - |
| M29 | Glucuronidation conjugation | C23H24O11 | 475.1253 | 1.4 | 7.64 | 429.2086, 299.0910, 271.0626, 179.0329, 119.0486 | 1.31755 | + | + | + | + | + |
| M30 | Glucuronidation conjugation | C23H24O11 | 475.1260 | 3.0 | 8.38 | 429.1939, 299.0971, 271.0566, 179.0331, 119.0488 | 1.35432 | + | + | + | + | + |
| M31 | Glucose conjugation | C23H26O10 | 461.1438 | −3.3 | 6.38 | 443.1360, 299.0894, 285.1122, 205.0585, 179.0306, 119.0515 | 1.32317 | - | - | + | + | - |
| M32 | Glucose conjugation | C23H26O10 | 461.1441 | −2.7 | 6.40 | 443.1351, 299.0889, 285.1121, 205.0575, 179.0336, 119.0525 | 1.7964 | - | - | + | + | - |
| M33 | Glucose conjugation | C23H26O10 | 461.1452 | −0.3 | 7.03 | 443.1353, 299.0874, 285.1134, 205.0565, 179.0332, 119.0521 | 1.83317 | - | - | + | + | - |
| M34 | Sulfate conjugation | C17H16O8S | 379.0495 | 0.4 | 6.57 | 374.0495, 299.0914, 205.0488, 179.0336, 119.0493 | 1.25962 | - | + | - | + | - |
| M35 | Sulfate conjugation | C17H16O8S | 379.0500 | 1.8 | 7.50 | 374.0500, 299.0879, 205.0476, 179.0324, 119.0485 | 1.73285 | - | - | + | + | - |
| M36 | Sulfate conjugation | C17H16O8S | 379.0495 | 0.6 | 8.26 | 374.0495, 299.0897, 205.0476, 179.0334, 119.0487 | 1.76962 | + | - | - | + | - |
| M37 | Methylation | C18H18O5 | 313.1086 | 1.7 | 8.09 | 298.0839, 271.0984, 269.1748, 219.0216, 193.0147, 119.0485 | 3.40562 | - | + | + | + | - |
| M38 | Methylation | C18H18O5 | 313.1078 | −1.1 | 16.84 | 298.0847, 271.0974,269.1737, 219.0223, 193.0156, 119.0489 | 3.91562 | - | - | + | + | - |
| M39 | Methylation | C18H18O5 | 313.1079 | −0.7 | 11.44 | 298.0841, 269.1763, 267.1589, 179.0338, 133.0296 | 3.87885 | + | - | + | + | - |
| M40 | N-acetylation | C19H18O6 | 341.1029 | −0.6 | 11.53 | 323.1861, 297.2049, 295.1910, 205.0479, 179.0351, 135.0442 | 3.30885 | - | - | + | + | - |
| M41 | N-acetylcysteine conjugation | C22H23NO8S | 460.1062 | −2.2 | 9.03 | 297.0769, 177.0185, 162.0226, 119.0497 | −0.41386 | - | + | + | + | - |
| M42 | N-acetylcysteine conjugation | C22H23NO8S | 460.1061 | −2.4 | 9.50 | 297.0779, 177.0179, 162.0231, 119.0487 | −0.41386 | - | - | + | + | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, J.; Ma, Y.; Liang, C.; Wang, H.; Sun, Y.; Zhang, L.; Jia, Q. A Complete Study of Farrerol Metabolites Produced In Vivo and In Vitro. Molecules 2019, 24, 3470. https://doi.org/10.3390/molecules24193470
Yin J, Ma Y, Liang C, Wang H, Sun Y, Zhang L, Jia Q. A Complete Study of Farrerol Metabolites Produced In Vivo and In Vitro. Molecules. 2019; 24(19):3470. https://doi.org/10.3390/molecules24193470
Chicago/Turabian StyleYin, Jintuo, Yinling Ma, Caijuan Liang, Hairong Wang, Yupeng Sun, Lantong Zhang, and Qingzhong Jia. 2019. "A Complete Study of Farrerol Metabolites Produced In Vivo and In Vitro" Molecules 24, no. 19: 3470. https://doi.org/10.3390/molecules24193470
APA StyleYin, J., Ma, Y., Liang, C., Wang, H., Sun, Y., Zhang, L., & Jia, Q. (2019). A Complete Study of Farrerol Metabolites Produced In Vivo and In Vitro. Molecules, 24(19), 3470. https://doi.org/10.3390/molecules24193470