
Assessing Parameter Suitability for the Strength Evaluation of Intramolecular Resonance Assisted Hydrogen Bonding in o-Carbonyl Hydroquinones

 ,
,
Abstract
:1. Introduction
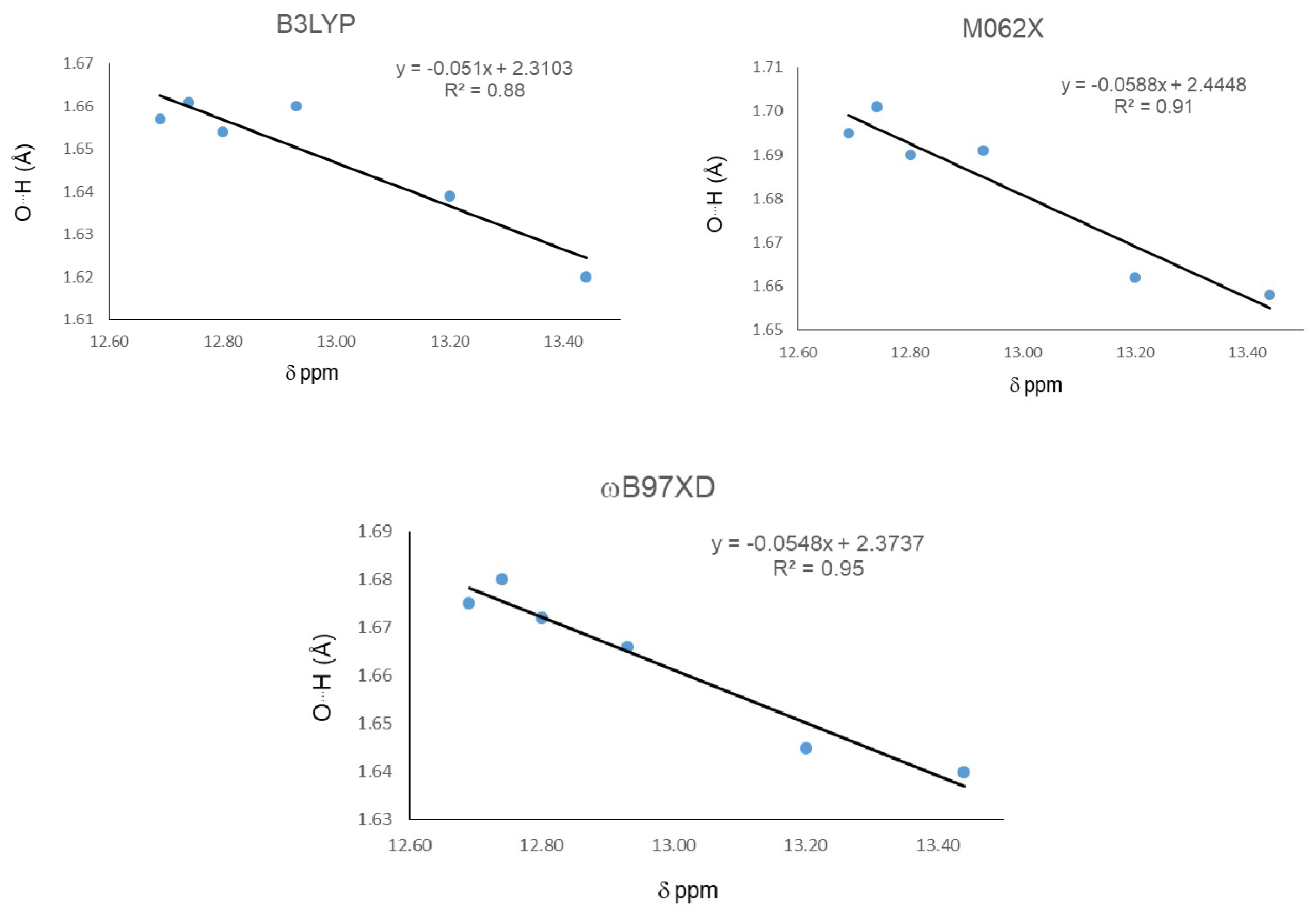
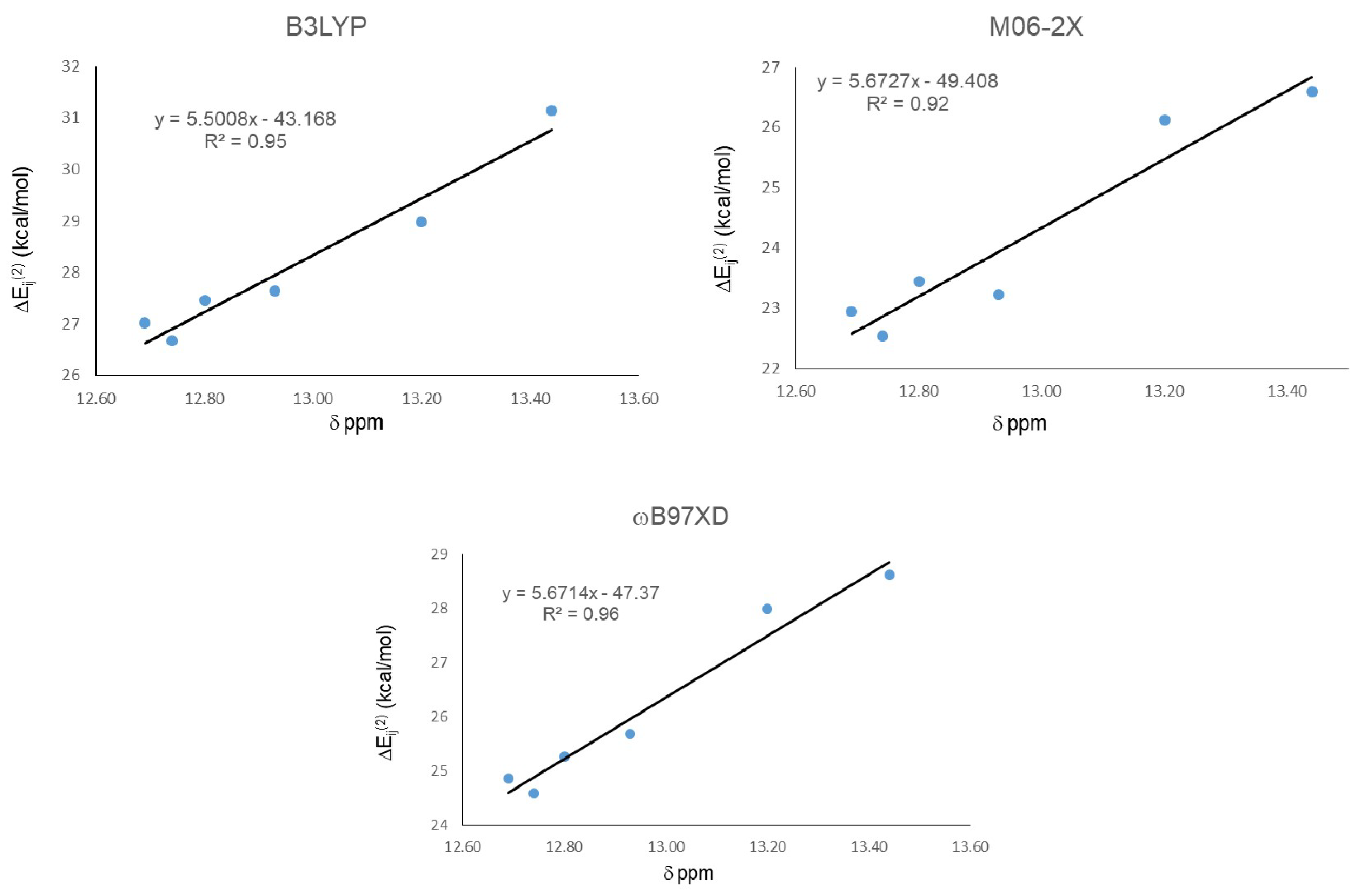
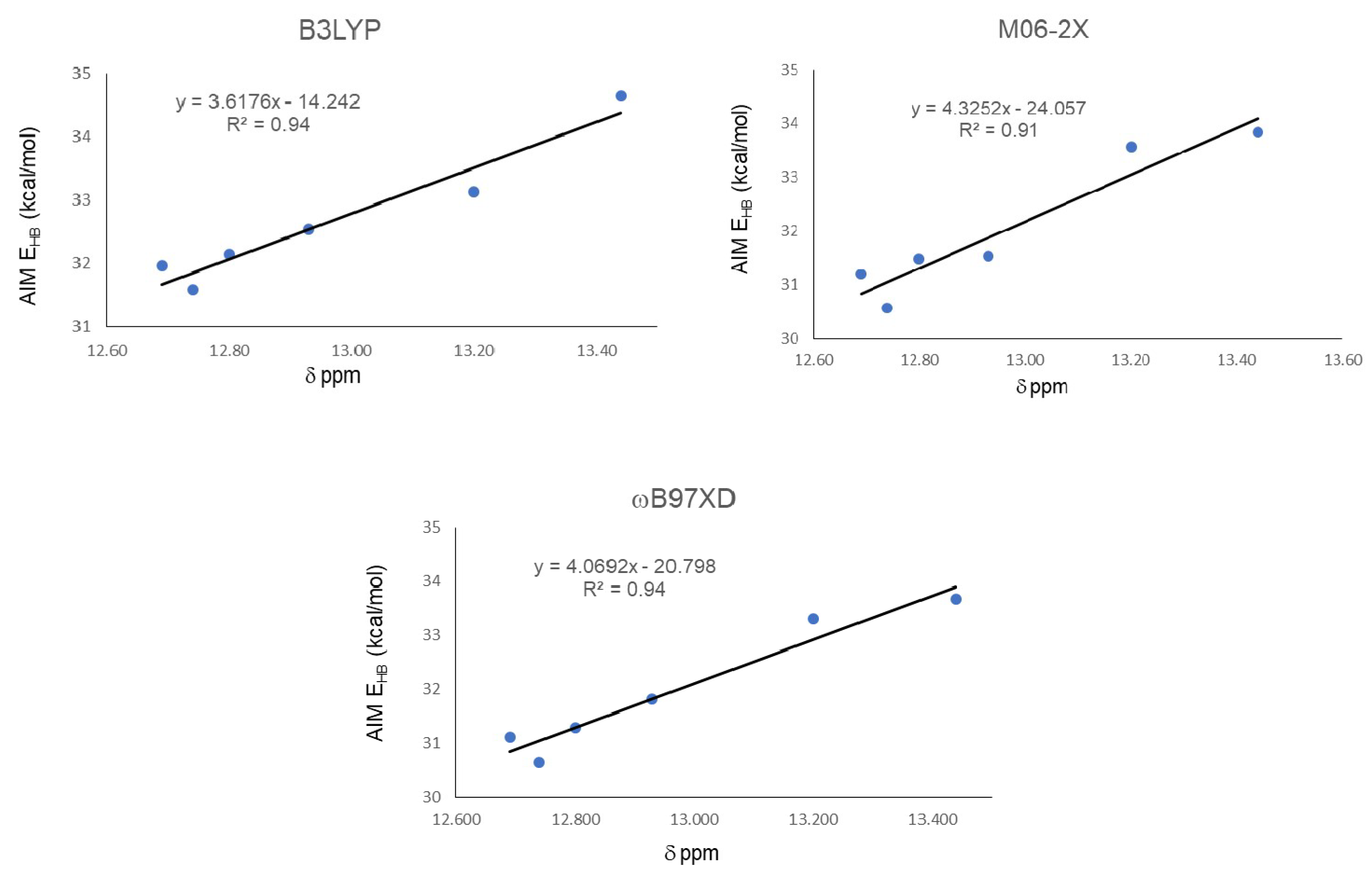
2. Results
3. Materials and Methods
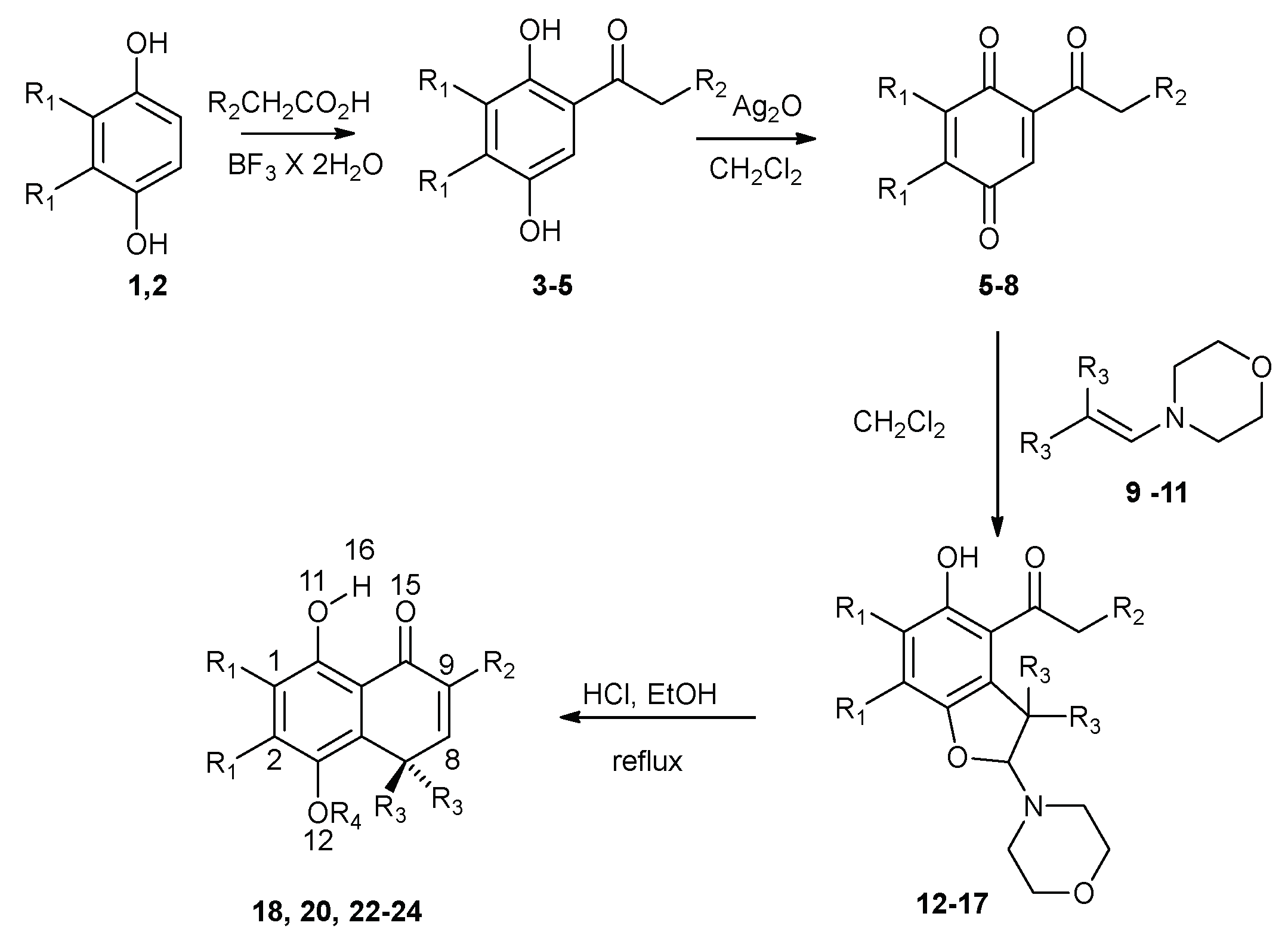
3.1. Synthetic Procedures
3.1.1. 1-(5-Hydroxy-3,3,6,7-tetramethyl-2-morpholino-2,3-dihydrobenzofuran-4-yl)ethan-1-one (14)
3.1.2. 5,8-Dihydroxy-4,4,6,7-tetramethylnaphthalen-1(4H)-one (21)
3.1.3. 8-Hydroxy-5-methoxy-4,4,6,7-tetramethylnaphthalen-1(4H)-one (22)
3.2. Computational Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Pairas, G.N.; Tsoungas, P.G. H-Bond: The Chemistry-Biology H-Bridge. Chemistryselect 2016, 1, 4520–4532. [Google Scholar] [CrossRef]
- Giordanetto, F.; Tyrchan, C.; Ulander, J. Intramolecular Hydrogen Bond Expectations in Medicinal Chemistry. ACS Med. Chem. Lett. 2017, 8, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Rezai, T.; Bock, J.E.; Zhou, M.V.; Kalyanaraman, C.; Lokey, R.S.; Jacobson, M.P. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: Successful in silico prediction of the relative permeabilities of cyclic peptides. J. Am. Chem. Soc. 2006, 128, 14073–14080. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Millan, D.S.; Perez, M.; Wakenhut, F.; Whitlock, G.A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Medchemcomm 2011, 2, 669–674. [Google Scholar] [CrossRef]
- Caron, G.; Vallaro, M.; Ermondi, G. High throughput methods to measure the propensity of compounds to form intramolecular hydrogen bonding. Medchemcomm 2017, 8, 1143–1151. [Google Scholar] [CrossRef]
- Afonin, A.V.; Sterkhova, I.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular bifurcated (three-centered) hydrogen bond by X-ray, IR and H-1 NMR spectroscopy, and QTAIM calculations. J. Mol. Struct. 2018, 1163, 185–196. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: An atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys. Chem. Chem. Phys. 2014, 16, 22935–22952. [Google Scholar] [CrossRef]
- Tang, S.S.; Tsona, N.T.; Du, L. Ring-Size Effects on the Stability and Spectral Shifts of Hydrogen Bonded Cyclic Ethers Complexes. Sci. Rep.-UK 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Bai, F.Q.; Wu, Y.; Zhang, H.X. Does the molecular structure of CaH2 affect the dihydrogen bonding in CaH2 center dot center dot center dot HY (Y = CH3, C2H3, C2H, CN, and NC) complexes? A quantum chemistry study using MP2 and B3LYP methods. Sci. China Chem. 2012, 55, 262–269. [Google Scholar] [CrossRef]
- Martinez-Cifuentes, M.; Cardona, W.; Saitz, C.; Weiss-Lopez, B.; Araya-Maturana, R. A Study about Regioisomeric Hydroquinones with Multiple Intramolecular Hydrogen Bonding. Molecules 2017, 22, 593. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Munoz, F.; Cordova-Delgado, M.; Ramirez, M.P.; Pena-Ahumada, B.; Rios, M.; Cruz, P.; Ahumada-Castro, U.; Bustos, G.; Silva-Pavez, E.; et al. FR58P1a; a new uncoupler of OXPHOS that inhibits migration in triple-negative breast cancer cells via Sirt1/AMPK/beta 1-integrin pathway. Sci. Rep.-UK 2018, 8, 16. [Google Scholar] [CrossRef]
- Urra, F.A.; Córdova-Delgado, M.; Lapier, M.; Orellana-Manzano, A.; Acevedo-Arévalo, L.; Pessoa-Mahana, H.; González-Vivanco, J.M.; Martínez-Cifuentes, M.; Ramírez-Rodróguez, O.; Millas-Vargas, J.P.; et al. Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol. Appl. Pharmacol. 2016, 291, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Martínez-Cifuentes, M.; Pavani, M.; Lapier, M.; Jaña-Prado, F.; Parra, E.; Maya, J.D.; Pessoa-Mahana, H.; Ferreira, J.; Araya-Maturana, R. An ortho-carbonyl substituted hydroquinone derivative is an anticancer agent that acts by inhibiting mitochondrial bioenergetics and by inducing G2/M-phase arrest in mammary adenocarcinoma TA3. Toxicol. Appl. Pharmacol. 2013, 267, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, M.; Araya-Maturana, R.; Palomo, I.; Fuentes, E. Platelet mitochondrial dysfunction and mitochondria-targeted quinone-and hydroquinone-derivatives: Review on new strategy of antiplatelet activity. Biochem. Pharmacol. 2018, 156, 215–222. [Google Scholar] [CrossRef]
- Dobado, J.A.; Gomez-Tamayo, J.C.; Calvo-Flores, F.G.; Martinez-Garcia, H.; Cardona, W.; Weiss-Lopez, B.; Ramirez-Rodriguez, O.; Pessoa-Mahana, H.; Araya-Maturana, R. NMR assignment in regioisomeric hydroquinones. Magn. Reson. Chem. 2011, 49, 358–365. [Google Scholar] [CrossRef]
- Salazar, R.; Vidal, J.; Martínez-Cifuentes, M.; Araya-Maturana, R.; Ramírez-Rodríguez, O. Electrochemical characterization of hydroquinone derivatives with different substituents in acetonitrile. New J. Chem. 2015, 39, 1237–1246. [Google Scholar] [CrossRef]
- Martinez-Cifuentes, M.; Salazar, R.; Escobar, C.A.; Weiss-Lopez, B.E.; Santos, L.S.; Araya-Maturana, R. Correlating experimental electrochemistry and theoretical calculations in 2’-hydroxy chalcones: The role of the intramolecular hydrogen bond. RSC Adv. 2015, 5, 50929–50937. [Google Scholar] [CrossRef]
- Rodriguez, J.; Olea-Azar, C.; Cavieres, C.; Norambuena, E.; Delgado-Castro, T.; Soto-Delgado, J.; Araya-Maturana, R. Antioxidant properties and free radical-scavenging reactivity of a family of hydroxynaphthalenones and dihydroxyanthracenones. Bioorgan. Med. Chem. 2007, 15, 7058–7065. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Chen, B.; Xie, H.; He, Y.H.; Zhong, D.W.; Chen, D.F. Antioxidant Structure-Activity Relationship Analysis of Five Dihydrochalcones. Molecules 2018, 23, 1162. [Google Scholar] [CrossRef] [PubMed]
- Lown, J.W. Molecular mechanisms of action of anticancer agents involving free radical intermediates. Adv. Free Radic. Biol. Med. 1985, 1, 225–264. [Google Scholar] [CrossRef]
- Pedroza, D.A.; De Leon, F.; Varela-Ramirez, A.; Lema, C.; Aguilera, R.J.; Mito, S. The cytotoxic effect of 2-acylated-1,4-naphthohydroquinones on leukemia/lymphoma cells. Bioorg. Med. Chem. 2014, 22, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.T.; Li, Z.L.; Wu, J.Y.; Lu, F.J.; Chen, C.H. An Oxidative Stress Mechanism of Shikonin in Human Glioma Cells. PLoS ONE 2014, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Loya, S.; Bakhanashvili, M.; Kashman, Y.; Hizi, A. Peyssonol-A and Peyssonal-B, 2 novel inhibitors of the reverse transcriptases of human-immunodeficiency-virus type-1 and type-2. Arch. Biochem. Biophys. 1995, 316, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef] [Green Version]
- Castro, C.G.; Santos, J.G.; Valcarcel, J.C.; Valderrama, J.A. Kinetic study of the acid-catalyzed rearrangement of 4-acetyl-3,3-dimethyl-5-hydroxy-2-morpholino-2,3-dihydrobenzo[b]furan. J. Org. Chem. 1983, 48, 3026–3029. [Google Scholar] [CrossRef]
- Araya-Maturana, R.; Cassels, B.K.; Delgado-Castro, T.; Valderrama, J.A.; Weiss-Lopez, B.E. Regioselectivity in the Diels-Alder reaction of 8,8-dimethylnaphthalene-1,4,5(8H)-trione with 2,4-hexadien-1-ol. Tetrahedron 1999, 55, 637–648. [Google Scholar] [CrossRef]
- Viglianisi, C.; Bartolozzi, M.G.; Pedulli, G.F.; Amorati, R.; Menichetti, S. Optimization of the Antioxidant Activity of Hydroxy-Substituted 4-Thiaflavanes: A Proof-of-Concept Study. Chem. Eur. J. 2011, 17, 12396–12404. [Google Scholar] [CrossRef]
- Araya-Maturana, R.; Delgado-Castro, T.; Garate, M.; Ferreira, J.; Pavani, M.; Pessoa-Mahana, H.; Cassels, B.K. Effects of 4,4-dimethyl-5,8-dihydroxynaphtalene-1-one and 4,4-dimethyl-5,8-dihydroxytetralone derivatives on tumor cell respiration. Bioorgan. Med. Chem. 2002, 10, 3057–3060. [Google Scholar] [CrossRef]
- Cassis, R.; Scholz, M.; Tapia, R.; Valderrama, J.A. Dienone-phenol rearrangement of naphthalenetriones-a route to 10-acetoxy-5,6,7,8-tetrahydrophenanthrene-1,4-diones. J. Chem. Soc. Perk. T 1987, 2855–2859. [Google Scholar] [CrossRef]
- Almodovar, I.; Ramirez-Rodriguez, O.; Barriga, A.; Rezende, M.C.; Araya-Maturana, R. Electrospray ionization mass spectrometric fragmentation of hydroquinone derivatives. Rapid. Commun. Mass. Spectrom. 2011, 25, 370–378. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond, Recent Development in Theory and Experiment; North Holland Publ. Co.: Amsterdam, The Netherland, 1976. [Google Scholar]
- Jablonski, M. Full vs. constrain geometry optimization in the open-closed method in estimating the energy of intramolecular charge-inverted hydrogen bonds. Chem. Phys. 2010, 376, 76–83. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H center dot center dot center dot F-Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D. Energy of Intramolecular Hydrogen Bonding in ortho-Hydroxybenzaldehydes, Phenones and Quinones. Transfer of Aromaticity from ipso-Benzene Ring to the Enol System(s). Molecules 2017, 22, 481. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Biegler-Konig, F.; Schonbohm, J. Update of the AIM2000-program for atoms in molecules. J. Comp. Chem. 2002, 23, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

| Hydroquinone | R1 | R2 | R3 | R4 |
| 18 [31] | H | H | Me | H |
| 19a [31] | H | H | Me | Me |
| 20 | Me | H | Me | H |
| 21b | Me | H | Me | Me |
| 22 [32] | H | Me | Me | H |
| 23 [32] | H | H | -CH2-(CH2)-CH2- | H |
| 24 [15,33] | H | H | Et | H |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
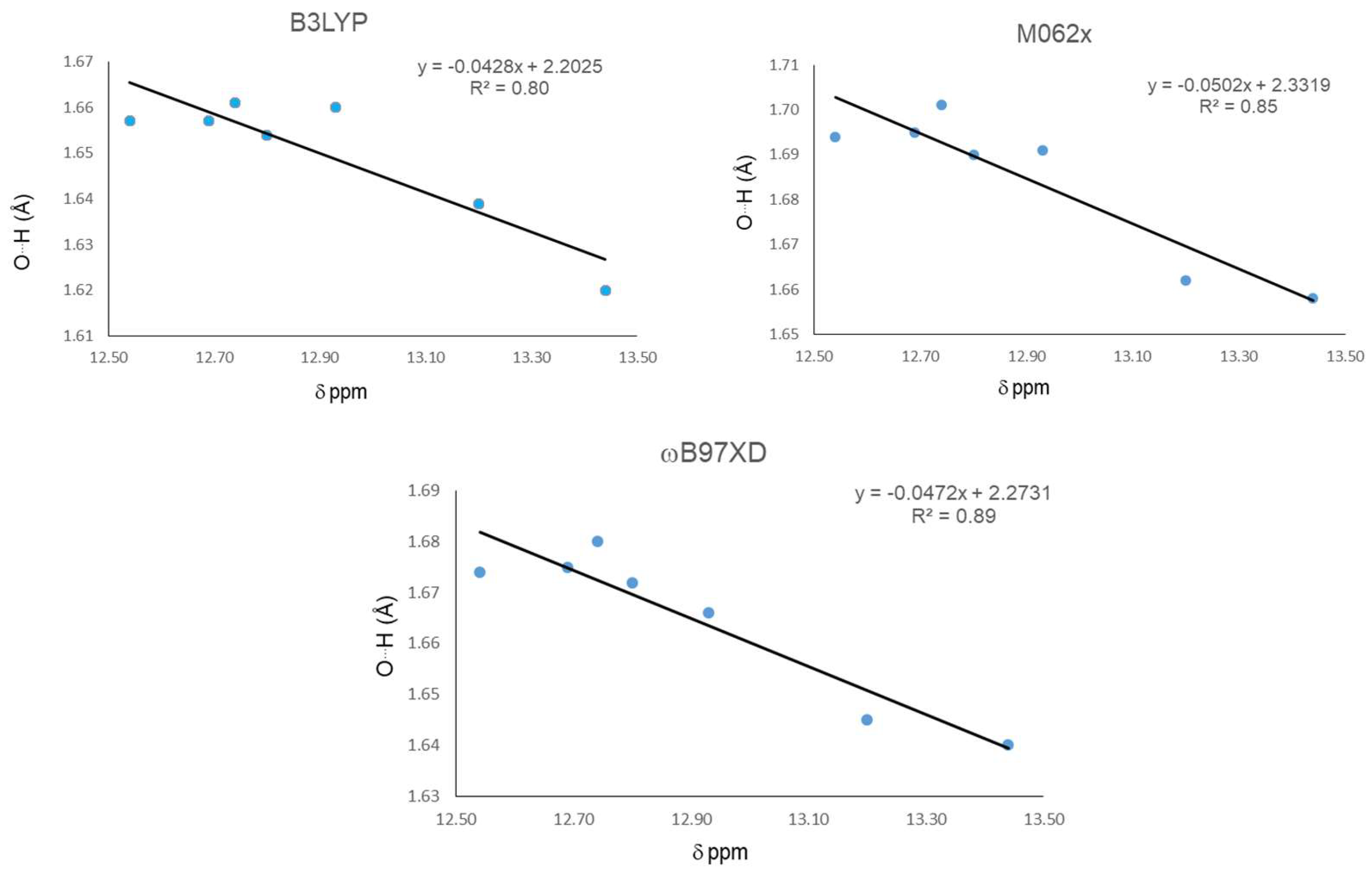
| Molecule | δ ppm | Distance (O15…H16) | Distance (O11-H16) | Angle (O15…H16-O11) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | M062x | ωB97XD | B3LYP | M062x | ωB97XD | B3LYP | M062x | ωB97XD | ||
| 18 | 12.540 | 1.657 | 1.694 | 1.674 | 0.99 | 0.98 | 0.982 | 148 | 146 | 147 |
| 19 | 12.690 | 1.657 | 1.695 | 1.675 | 0.99 | 0.98 | 0.981 | 148 | 146 | 147 |
| 20 | 13.200 | 1.639 | 1.662 | 1.645 | 0.992 | 0.983 | 0.984 | 149 | 147 | 148 |
| 21 | 13.440 | 1.620 | 1.658 | 1.640 | 0.994 | 0.984 | 0.985 | 149 | 148 | 149 |
| 22 | 12.930 | 1.660 | 1.691 | 1.666 | 0.99 | 0.98 | 0.982 | 148 | 146 | 147 |
| 23 | 12.740 | 1.661 | 1.701 | 1.68 | 0.99 | 0.98 | 0.982 | 148 | 146 | 147 |
| 24 | 12.800 | 1.654 | 1.69 | 1.672 | 0.99 | 0.981 | 0.982 | 148 | 146 | 147 |
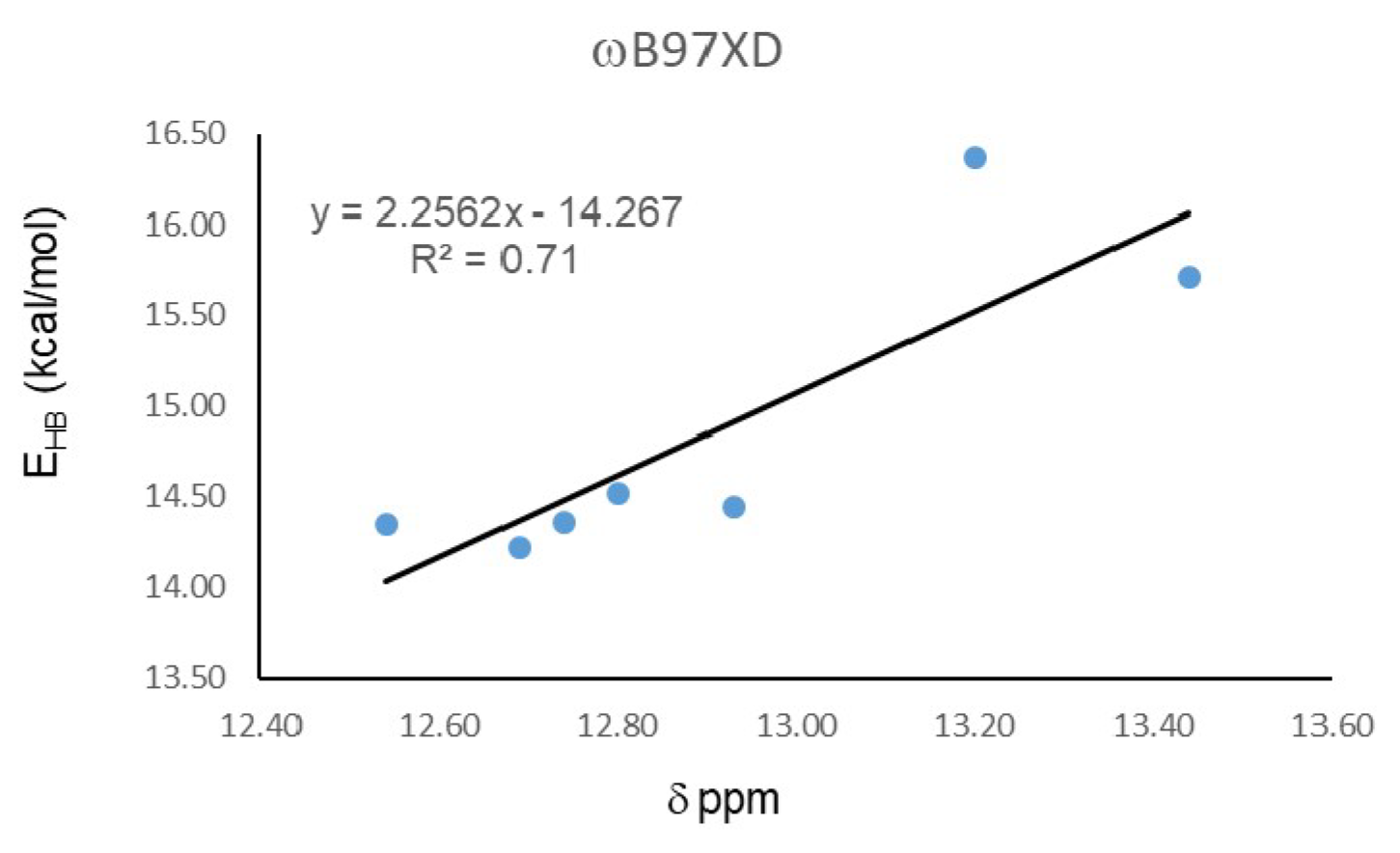
| Molecule | EHB |
|---|---|
| 18 | 14.34 |
| 19 | 14.22 |
| 20 | 16.37 |
| 21 | 15.71 |
| 22 | 14.44 |
| 23 | 14.36 |
| 24 | 14.52 |
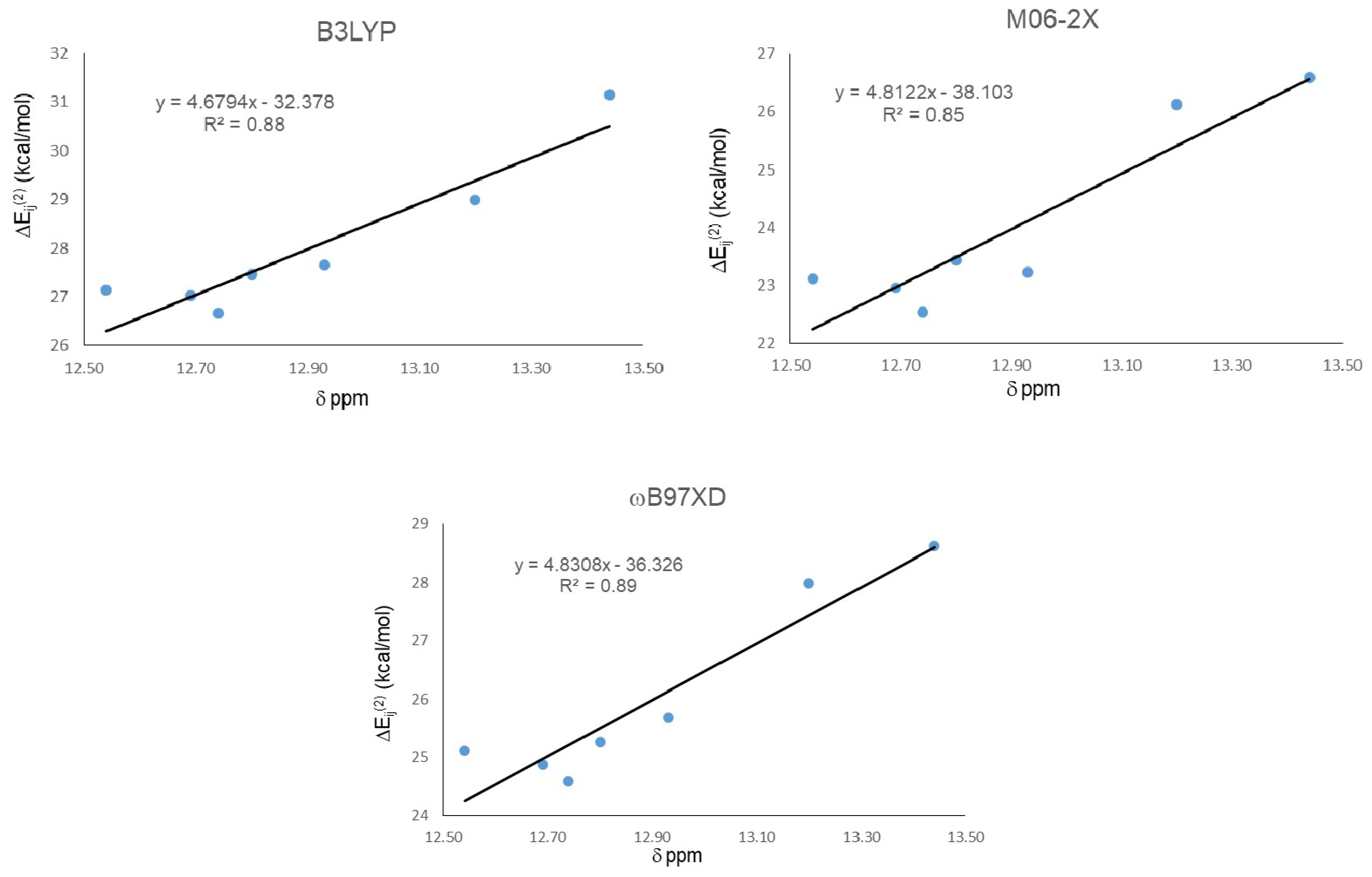
| Molecule | ΔEij(2) LP O15 → σ* O11-H16 | ||
|---|---|---|---|
| B3LYP | M06-2X | ωB97XD | |
| 18 | 27.14 | 23.12 | 25.11 |
| 19 | 27.02 | 22.95 | 24.87 |
| 20 | 28.99 | 26.13 | 27.99 |
| 21 | 31.15 | 26.6 | 28.62 |
| 22 | 27.65 | 23.23 | 25.68 |
| 23 | 26.67 | 22.54 | 24.59 |
| 24 | 27.47 | 23.44 | 25.27 |
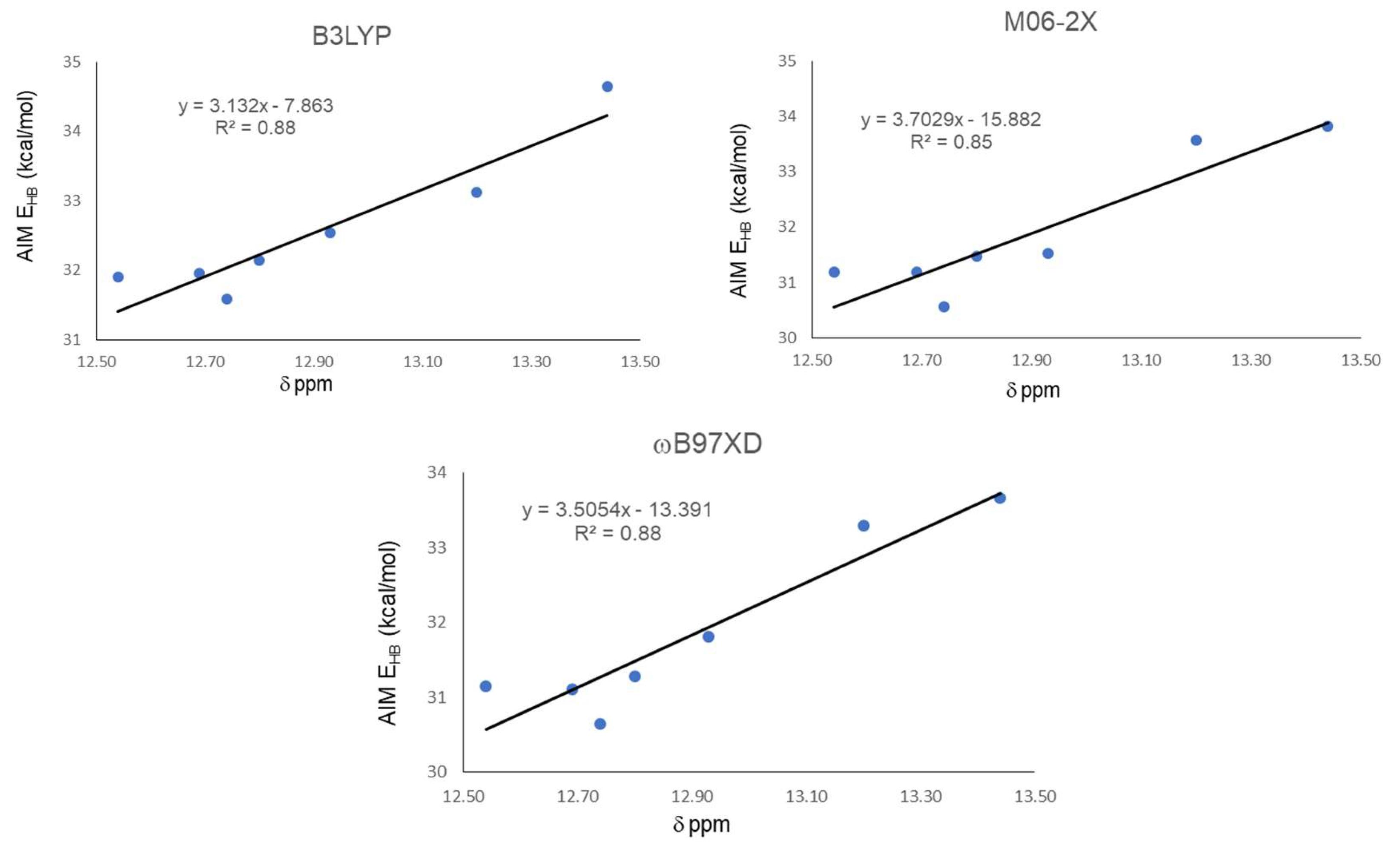
| Functional | Molecule | ρBCP | ∇2ρ | G | V | |V|/G | H | EHB |
|---|---|---|---|---|---|---|---|---|
| B3LYP | 18 | 0.0537 | −0.0377 | 0.0459 | −0.1013 | 2.2070 | −0.1472 | 31.91 |
| 19 | 0.0537 | −0.0378 | 0.0460 | −0.1015 | 2.2065 | −0.1475 | 31.96 | |
| 20 | 0.0560 | −0.0383 | 0.0478 | −0.1051 | 2.1987 | −0.1529 | 33.12 | |
| 21 | 0.0587 | −0.0392 | 0.0501 | −0.1100 | 2.1956 | −0.1601 | 34.65 | |
| 22 | 0.0546 | −0.0383 | 0.0469 | −0.1033 | 2.2025 | −0.1502 | 32.54 | |
| 23 | 0.0533 | −0.0374 | 0.0455 | −0.1003 | 2.2044 | −0.1457 | 31.59 | |
| 24 | 0.0541 | −0.0378 | 0.0463 | −0.1020 | 2.2030 | −0.1483 | 32.14 | |
| M06-2X | 18 | 0.0454 | −0.0408 | 0.0444 | −0.0990 | 2.2297 | −0.1434 | 31.19 |
| 19 | 0.0454 | −0.0409 | 0.0444 | −0.0990 | 2.2297 | −0.1434 | 31.19 | |
| 20 | 0.0490 | −0.0427 | 0.0479 | −0.1066 | 2.2255 | −0.1545 | 33.57 | |
| 21 | 0.0495 | −0.0428 | 0.0483 | −0.1074 | 2.2236 | −0.1557 | 33.83 | |
| 22 | 0.0458 | −0.0412 | 0.0449 | −0.1001 | 2.2294 | −0.1450 | 31.53 | |
| 23 | 0.0446 | −0.0402 | 0.0435 | −0.0970 | 2.2299 | −0.1405 | 30.57 | |
| 24 | 0.0459 | −0.0410 | 0.0448 | −0.0999 | 2.2299 | −0.1448 | 31.48 | |
| ωB97XD | 18 | 0.0510 | −0.0380 | 0.0447 | −0.0989 | 2.2125 | −0.1435 | 31.14 |
| 19 | 0.0508 | −0.0381 | 0.0446 | −0.0987 | 2.2130 | −0.1434 | 31.10 | |
| 20 | 0.0546 | −0.0394 | 0.0479 | −0.1057 | 2.2067 | −0.1536 | 33.30 | |
| 21 | 0.0553 | −0.0396 | 0.0485 | −0.1069 | 2.2041 | −0.1553 | 33.66 | |
| 22 | 0.0518 | −0.0386 | 0.0457 | −0.1010 | 2.2100 | −0.1466 | 31.81 | |
| 23 | 0.0502 | −0.0376 | 0.0439 | −0.0973 | 2.2164 | −0.1412 | 30.64 | |
| 24 | 0.0512 | −0.0381 | 0.0449 | −0.0993 | 2.2116 | −0.1442 | 31.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Monroy-Cárdenas, M.; Millas-Vargas, J.P.; Weiss-López, B.E.; Araya-Maturana, R. Assessing Parameter Suitability for the Strength Evaluation of Intramolecular Resonance Assisted Hydrogen Bonding in o-Carbonyl Hydroquinones. Molecules 2019, 24, 280. https://doi.org/10.3390/molecules24020280
Martínez-Cifuentes M, Monroy-Cárdenas M, Millas-Vargas JP, Weiss-López BE, Araya-Maturana R. Assessing Parameter Suitability for the Strength Evaluation of Intramolecular Resonance Assisted Hydrogen Bonding in o-Carbonyl Hydroquinones. Molecules. 2019; 24(2):280. https://doi.org/10.3390/molecules24020280
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Matías Monroy-Cárdenas, Juan Pablo Millas-Vargas, Boris E. Weiss-López, and Ramiro Araya-Maturana. 2019. "Assessing Parameter Suitability for the Strength Evaluation of Intramolecular Resonance Assisted Hydrogen Bonding in o-Carbonyl Hydroquinones" Molecules 24, no. 2: 280. https://doi.org/10.3390/molecules24020280
APA StyleMartínez-Cifuentes, M., Monroy-Cárdenas, M., Millas-Vargas, J. P., Weiss-López, B. E., & Araya-Maturana, R. (2019). Assessing Parameter Suitability for the Strength Evaluation of Intramolecular Resonance Assisted Hydrogen Bonding in o-Carbonyl Hydroquinones. Molecules, 24(2), 280. https://doi.org/10.3390/molecules24020280