Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds





Abstract
:
1. Introduction
2. Results
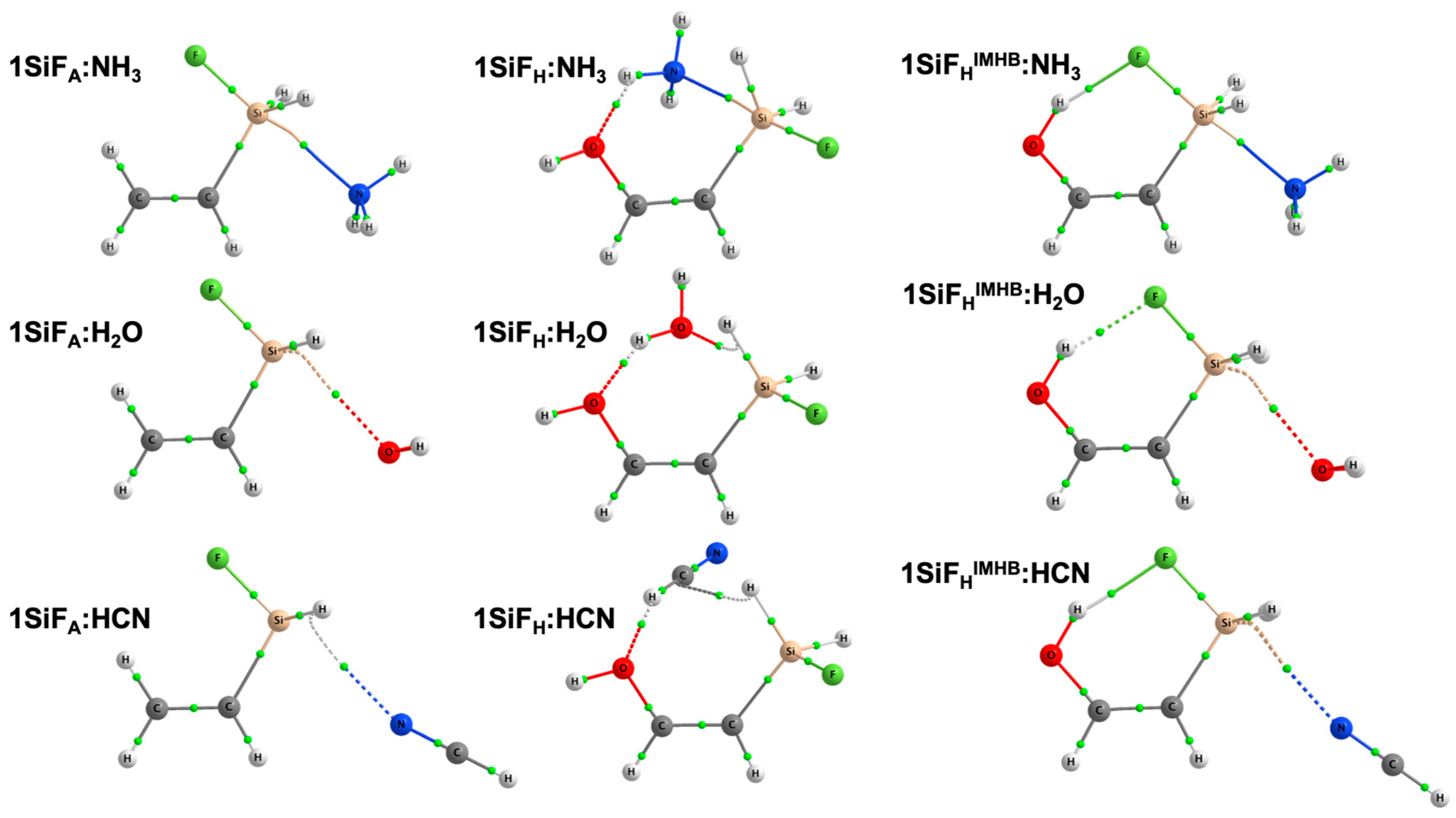
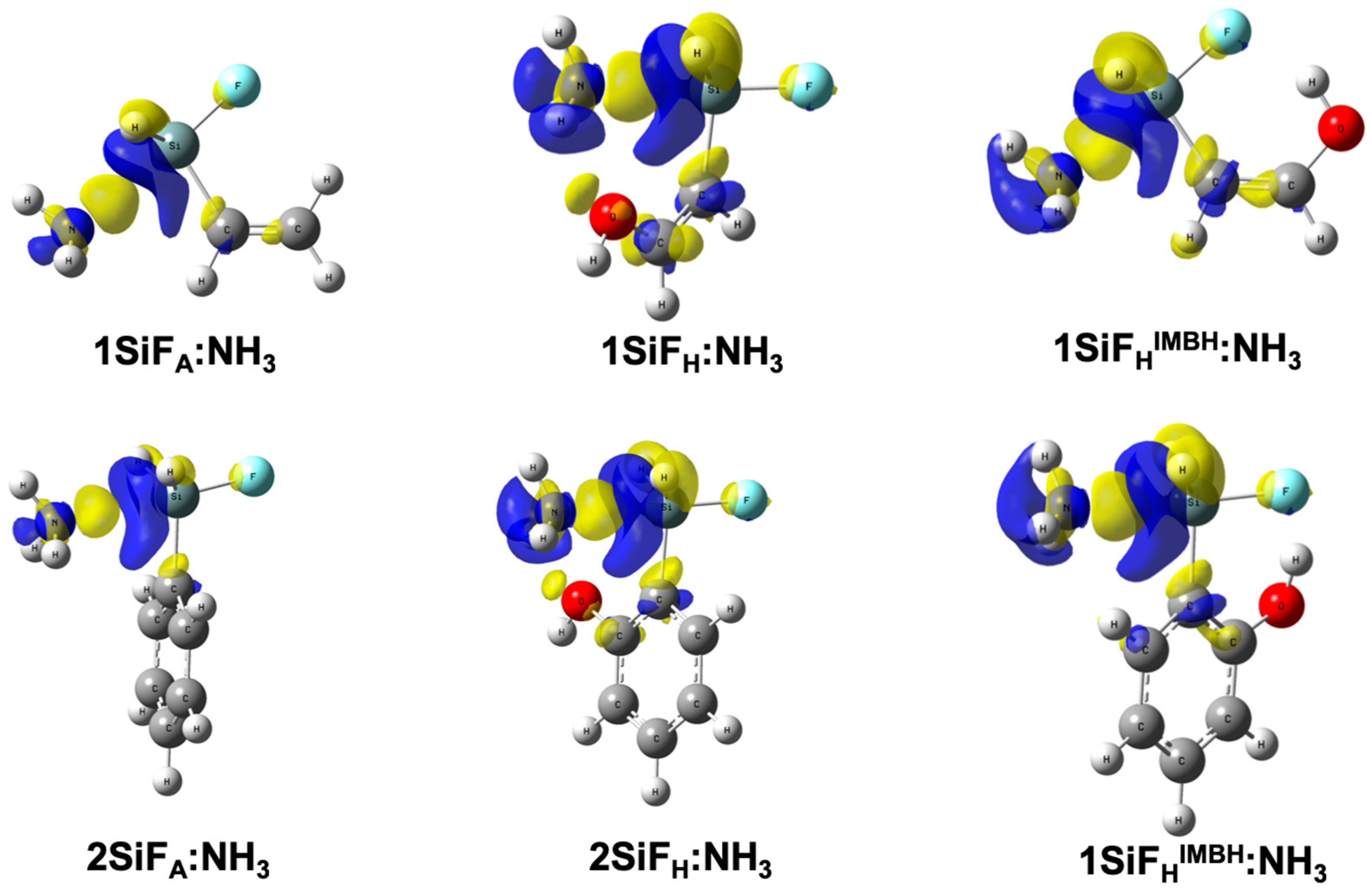
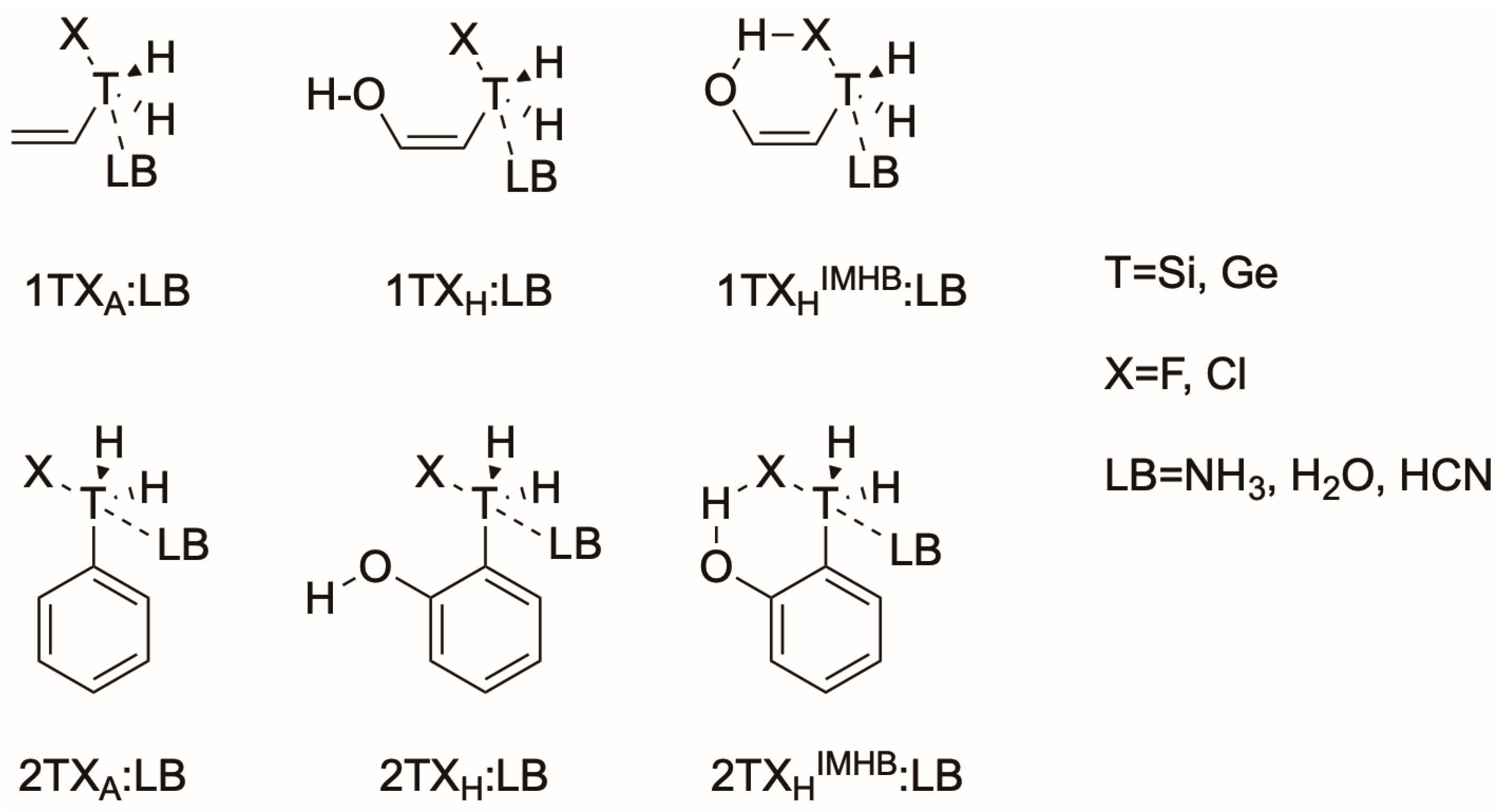
2.1. Allylfluorotetrel Derivatives: Effect of the Lewis Bases (1TF:LB)
2.2. Allylhalotetrel Derivatives: Effect of the Halogen (1TX:NH3)
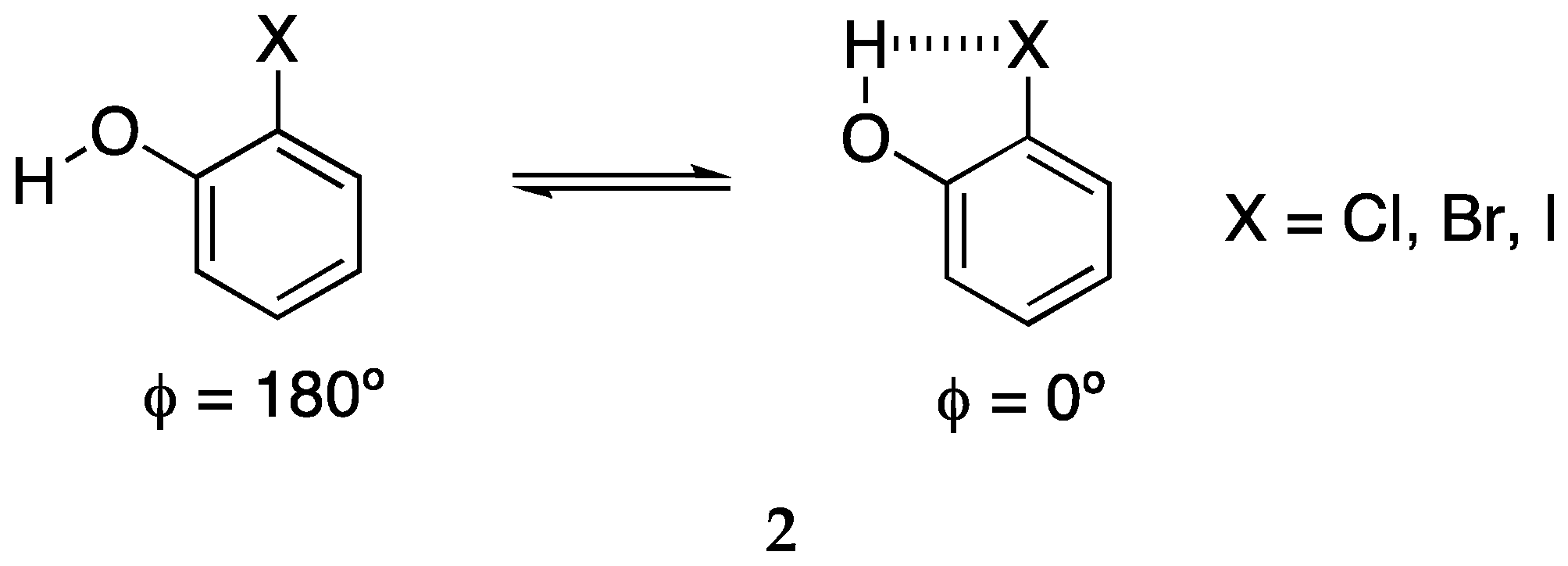
2.3. Phenyl Halogen Tetrel Derivatives: Effect of the Backbone (2TX:NH3)
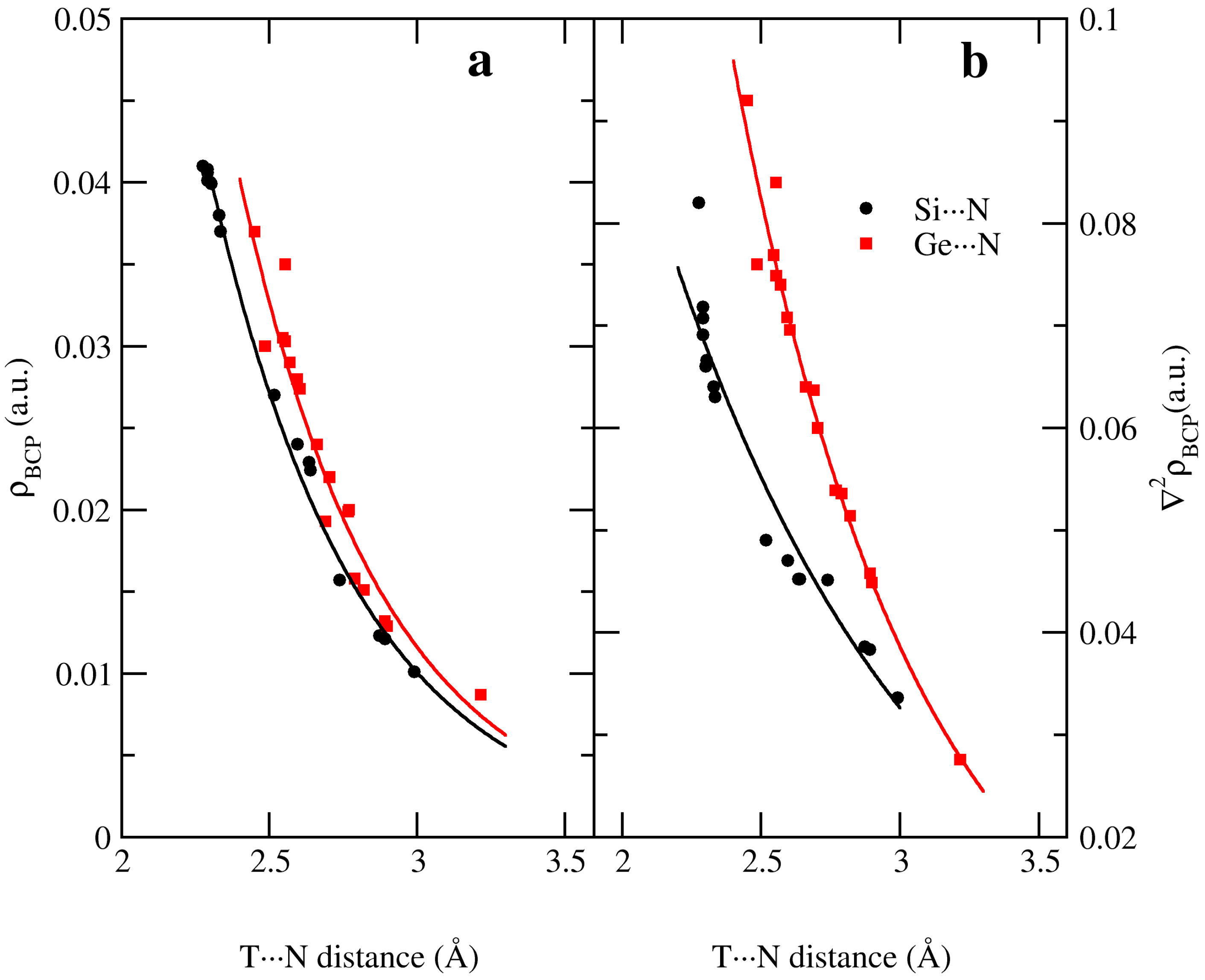
2.4. Electron Density Properties
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lehn, J.M. Supramolecular Chemistry Concepts and Perspectives; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Sensitivity of noncovalent bonds to intermolecular separation: Hydrogen, halogen, chalcogen, and pnicogen bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Biella, S. Halogen Bonding: Fundamentals and Applications; Springer: Berlin, Germany, 2008. [Google Scholar]
- Rozas, I.; Alkorta, I.; Elguero, J. Field effects on dihydrogen bonded systems. Chem. Phys. Lett. 1997, 275, 423–428. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Inverse Hydrogen-Bonded Complexes. J. Phys. Chem. A 1997, 101, 4236–4244. [Google Scholar] [CrossRef]
- Scheiner, S. Effects of substituents upon the P···N noncovalent interaction: The limits of its strength. J. Phys. Chem. A 2011, 115, 11202–11209. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1,2-dicarba-closo-dodecaboranes with short P⋯P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J.; Sánchez-Sanz, G. Lone-pair hole on P: P···N pnicogen bonds assisted by halogen bonds. J. Phys. Chem. A 2017, 121, 1362–1370. [Google Scholar] [CrossRef]
- Sanchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Modulating intramolecular P···N pnictogen interactions. Phys. Chem. Chem. Phys. 2016, 18, 9148–9160. [Google Scholar] [CrossRef]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Halogen, chalcogen and pnictogen interactions in (XNO2)2 homodimers (X = F, Cl, Br, I). New J. Chem. 2015, 39, 6791–6802. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular weak interactions in HTeXH dimers (X=O, S, Se, Te): Hydrogen bonds, chalcogen–chalcogen contacts and chiral discrimination. Chem. Phys. Chem. 2012, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Alkorta, I.; Elguero, J. Theoretical study of the HXYH dimers (X, Y = O, S, Se). Hydrogen bonding and chalcogen–chalcogen interactions. Mol. Phys. 2011, 109, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Azofra, L.; Alkorta, I.; Scheiner, S. Noncovalent interactions in dimers and trimers of SO3 and CO. Theor. Chem. Acc. 2014, 133, 1–8. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Resonance-assisted intramolecular chalcogen–chalcogen interactions? Chem. Eur. J. 2003, 9, 4548–4555. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C. Improvement of anion transport systems by modulation of chalcogen interactions: The influence of solvent. J. Phys. Chem. A 2018, 122, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Significant evidence of C⋯O and C⋯C long-range contacts in several heterodimeric complexes of CO with CH3–X, should one refer to them as carbon and dicarbon bonds! Phys. Chem. Chem. Phys. 2014, 16, 17238–17252. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Murray, J.; Concha, M.; Lane, P.; Hobza, P.; Politzer, P. Blue shifts vs. red shifts in σ-hole bonding. J. Mol. Model. 2008, 14, 699–704. [Google Scholar] [CrossRef]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Buckingham, A.D.; Fowler, P.W. A model for the geometries of Van der Waals complexes. Can. J. Chem. 1985, 63, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C.; Millen, D.J. Angular geometries and other properties of hydrogen-bonded dimers: A simple electrostatic interpretation of the success of the electron-pair model. Chem. Soc. Rev. 1987, 16, 467–498. [Google Scholar] [CrossRef]
- Stone, A.J.; Price, S.L. Some new ideas in the theory of intermolecular forces: Anisotropic atom-atom potentials. J. Phys. Chem. 1988, 92, 3325–3335. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Burling, F.T.; Goldstein, B.M. Computational studies of nonbonded sulfur-oxygen and selenium-oxygen interactions in the thiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313–2320. [Google Scholar] [CrossRef]
- Price, S.L. Applications of realistic electrostatic modelling to molecules in complexes, solids and proteins. J. Chem. Soc. Faraday Trans. 1996, 92, 2997–3008. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The nature of Halogen⋅⋅⋅Halogen synthons: Crystallographic and theoretical studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Hennemann, M.; Murray, J.; Politzer, P.; Riley, K.; Clark, T. Polarization-induced σ-holes and hydrogen bonding. J. Mol. Model. 2012, 18, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Hunter, C.A.; Anderson, H.L. What is cooperativity? Angew. Chem. Int. Ed. 2009, 48, 7488–7499. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Riel, A.M.S.; Berryman, O.B. Solvatochromism and fluorescence response of a halogen bonding anion receptor. New J. Chem. 2018, 42, 10489–10492. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.-C.C.; Scholfield, M.R.; Rowe, R.K.; Ford, M.C.; Alexander, A.T.; Mehl, R.A.; Ho, P.S. Increasing enzyme stability and activity through hydrogen bond-enhanced halogen bonds. Biochemistry 2018, 57, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Ośmiałowski, B.; Kolehmainen, E.; Gawinecki, R.; Kauppinen, R.; Koivukorpi, J.; Valkonen, A. NMR and quantum chemical studies on association of 2,6-bis(acylamino)pyridines with selected imides and 2,2′-dipyridylamine. Struct. Chem. 2010, 21, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B.J.; Kjaergaard, H.G. Are bond critical points really critical for hydrogen bonding? J. Chem. Theor. Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Bond paths between distant atoms do not necessarily indicate dominant interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal features of the electron density distribution in hydrogen-bonding regions: A comprehensive study involving H···X (X=H, C, N, O, F, S, Cl, π) interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 1998, 102, 9278–9285. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, energies, bonding, and NMR properties of pnicogen complexes H2XP:NXH2 (X = H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Blanco, F.; Elguero, J.; Dobado, J.A.; Ferrer, S.M.; Vidal, I. Carbon···Carbon weak interactions. J. Phys. Chem. A 2009, 113, 8387–8393. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croatica Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Aronld, W.D.; Oldfield, E. The chemical nature of hydrogen bonding in proteins via NMR: J-couplings, chemical shifts, and AIM theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of pnicogen bonding with the help of vibrational spectroscopy—The missing link between theory and experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Theoretical study of cyanophosphines: Pnicogen vs. dipole-dipole interactions. Comput. Theor. Chem. 2015, 1053, 305–314. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intramolecular pnicogen interactions in phosphorus and arsenic analogues of proton sponges. Phys. Chem. Chem. Phys. 2014, 16, 15900–15909. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. Intramolecular pnicogen interactions in PHF-(CH2)n-PHF (n = 2–6) systems. Chem. Phys. Chem. 2013, 14, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Weak interactions between hypohalous acids and dimethylchalcogens. Phys. Chem. Chem. Phys. 2012, 14, 9880–9889. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jørgensen, P.; Taylor, P.R. Basis set convergence of the interaction energy of hydrogen-bonded complexes. J. Chem. Phys. 1999, 111, 9157–9167. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Claredon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Prentice-Hall: Manchester, UK, 2000. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| avdz a | avtz a | avqz a | CBSDT b | CBSTQ b | |
|---|---|---|---|---|---|
| 1SiFA:NH3 | −29.3 | −25.4 | −23.8 | −24.5 | −21.8 |
| 1SiFH:NH3 | −32.3 | −28.2 | −25.9 | −27.4 | −22.9 |
| 1SiFHIMHB:NH3 | −40.5 | −36.3 | −34.4 | −35.4 | −31.7 |
| Comp. | NH3 | H2O | HCN |
|---|---|---|---|
| 1SiFA:LB | 2.518 | 2.885 | 2.991 |
| 1SiFH:LB | 2.335 | 2.883 | 4.033 |
| 1SiFHIMHB:LB | 2.276 | 2.765 | 2.873 |
| 1GeFA:LB | 2.661 | 2.829 | 2.898 |
| 1GeFH:LB | 2.570 | 2.782 | 2.891 |
| 1GeFHIMHB:LB | 2.450 | 2.720 | 2.789 |
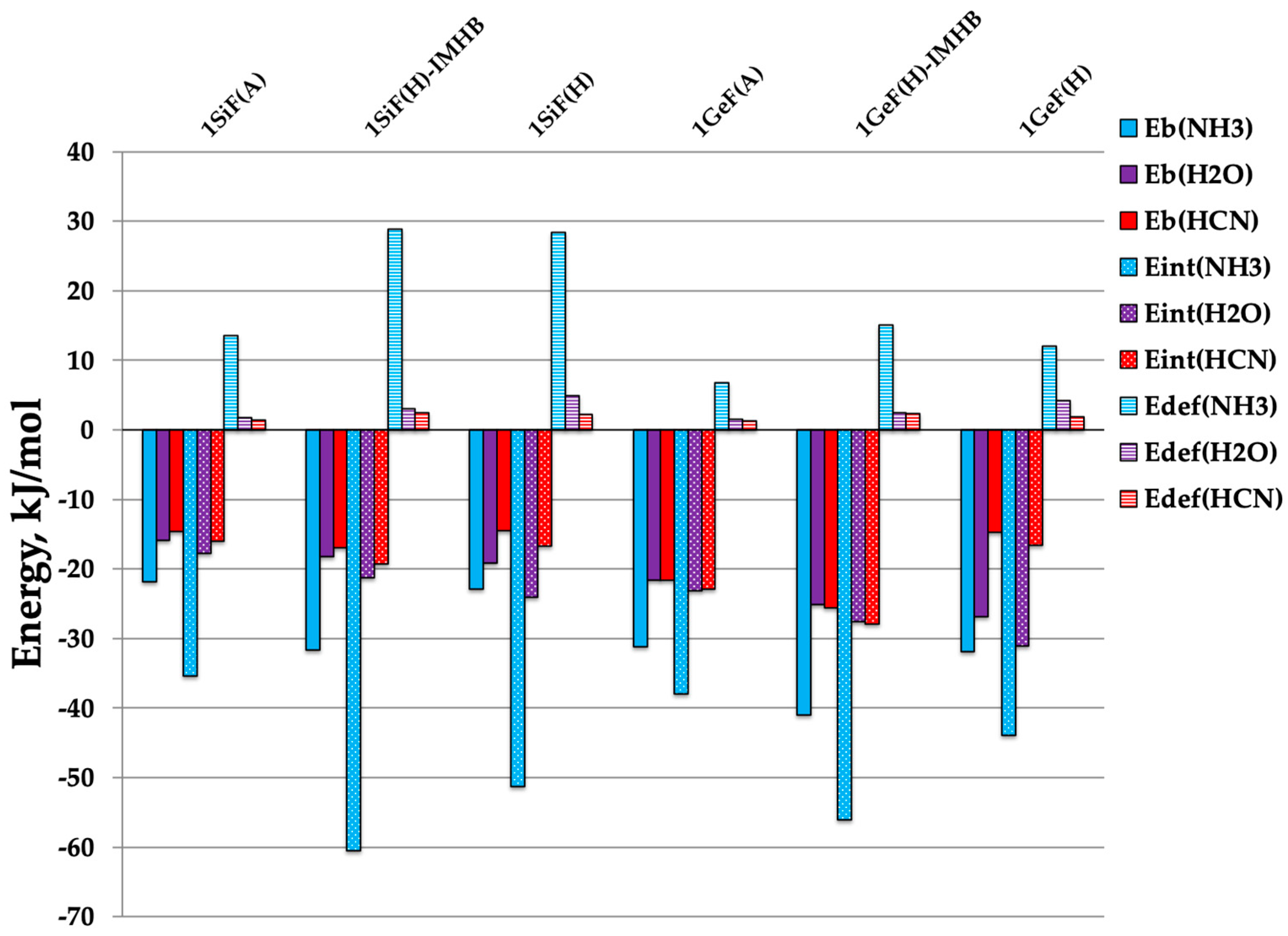
| Complex | Eb | Eint | Edef | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NH3 | H2O | HCN | NH3 | H2O | HCN | NH3 | H2O | HCN | |
| 1SiFA:NH3 | −21.8 | −15.9 | −14.6 | −35.2 | −17.7 | −16.1 | 13.6 | 1.8 | 1.4 |
| 1SiFH:NH3 | −22.9 | −19.2 | −14.5 | −51.3 | −24.1 | −16.7 | 28.4 | 4.9 | 2.2 |
| 1SiFHIMHB:NH3 | −31.7 | −18.2 | −16.9 | −60.6 | −21.2 | −19.2 | 28.8 | 3.0 | 2.4 |
| 1GeFA:NH3 | −31.2 | −21.6 | −21.6 | −38.0 | −23.1 | −22.9 | 6.8 | 1.5 | 1.3 |
| 1GeFH:NH3 | −31.9 | −26.9 | −14.7 | −43.9 | −31.1 | −16.6 | 12.0 | 4.2 | 1.9 |
| 1GeFHIMHB:NH3 | −41.0 | −25.1 | −25.6 | −56.1 | −27.6 | −27.9 | 15.1 | 2.4 | 2.3 |
| Eb | Eint | Edef | T···N | T-X | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl |
| 1SiXA:NH3 | −21.8 | −20.1 | −35.4 | −30.5 | 13.6 | 10.4 | 2.518 | 2.640 | 1.643 | 2.110 |
| 1SiXH:NH3 | −22.9 | −24.5 | −51.3 | −58.5 | 28.4 | 34.1 | 2.335 | 2.291 | 1.654 | 2.156 |
| 1SiXHIMHB:NH3 | −31.7 | −27.5 | −60.6 | −58.9 | 28.8 | 31.4 | 2.276 | 2.291 | 1.685 | 2.185 |
| 1GeXA:NH3 | −31.2 | −28.2 | −38.0 | −32.7 | 6.8 | 4.6 | 2.661 | 2.767 | 1.768 | 2.193 |
| 1GeXH:NH3 | −31.9 | −31.6 | −43.9 | −42.3 | 12.0 | 10.7 | 2.570 | 2.603 | 1.776 | 2.217 |
| 1GeXHIMHB:NH3 | −41.0 | −34.8 | −56.1 | −46.6 | 15.1 | 11.8 | 2.450 | 2.545 | 1.815 | 2.251 |
| Eb | Eint | Edef | T···N | T-X | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl |
| 2SiXA:NH3 | −21.5 | −21.5 | −30.99 | −30.78 | 9.5 | 9.2 | 2.596 | 2.635 | 1.638 | 2.111 |
| 2SiXH:NH3 | −23.7 | −28.7 | −59.13 | −61.07 | 33.9 | 33.2 | 2.330 | 2.305 | 1.652 | 2.150 |
| 2SiXHIMHB:NH3 | −25.2 | −27.9 | −55.27 | −59.79 | 31.6 | 31.1 | 2.301 | 2.291 | 1.677 | 2.176 |
| 2GeXA:NH3 | −29.0 | −27.8 | −34.45 | −31.73 | 5.4 | 3.9 | 2.703 | 2.771 | 1.766 | 2.194 |
| 2GeXH:NH3 | −32.1 | −31.3 | −55.44 | −48.18 | 15.3 | 13.3 | 2.553 | 2.594 | 1.776 | 2.214 |
| 2GeXHIMHB:NH3 | −40.1 | −34.9 | −47.60 | −44.99 | 15.5 | 13.7 | 2.485 | 2.554 | 1.809 | 2.244 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trujillo, C.; Alkorta, I.; Elguero, J.; Sánchez-Sanz, G. Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules 2019, 24, 308. https://doi.org/10.3390/molecules24020308
Trujillo C, Alkorta I, Elguero J, Sánchez-Sanz G. Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules. 2019; 24(2):308. https://doi.org/10.3390/molecules24020308
Chicago/Turabian StyleTrujillo, Cristina, Ibon Alkorta, José Elguero, and Goar Sánchez-Sanz. 2019. "Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds" Molecules 24, no. 2: 308. https://doi.org/10.3390/molecules24020308
APA StyleTrujillo, C., Alkorta, I., Elguero, J., & Sánchez-Sanz, G. (2019). Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules, 24(2), 308. https://doi.org/10.3390/molecules24020308