4.3. Synthesis
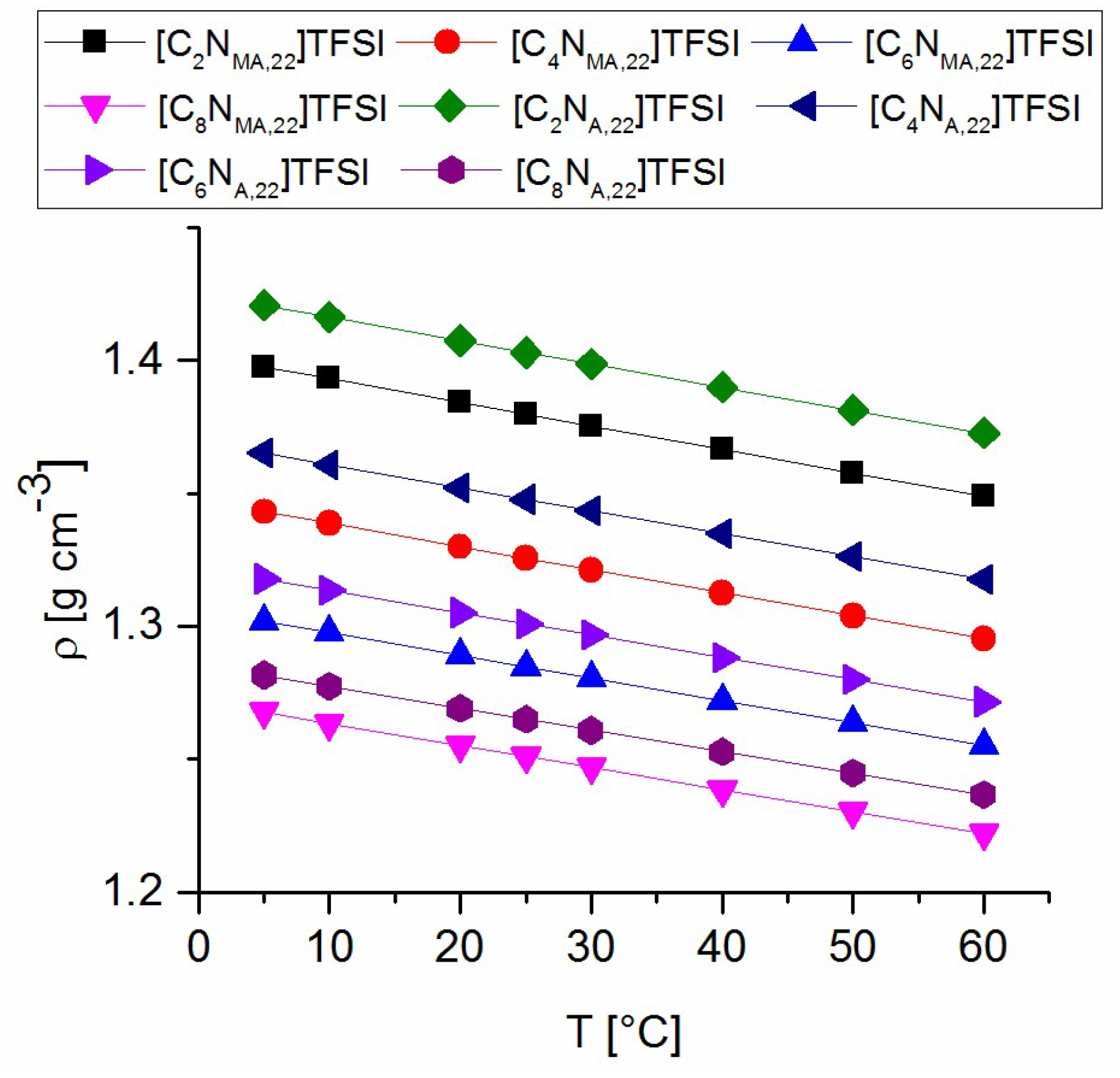
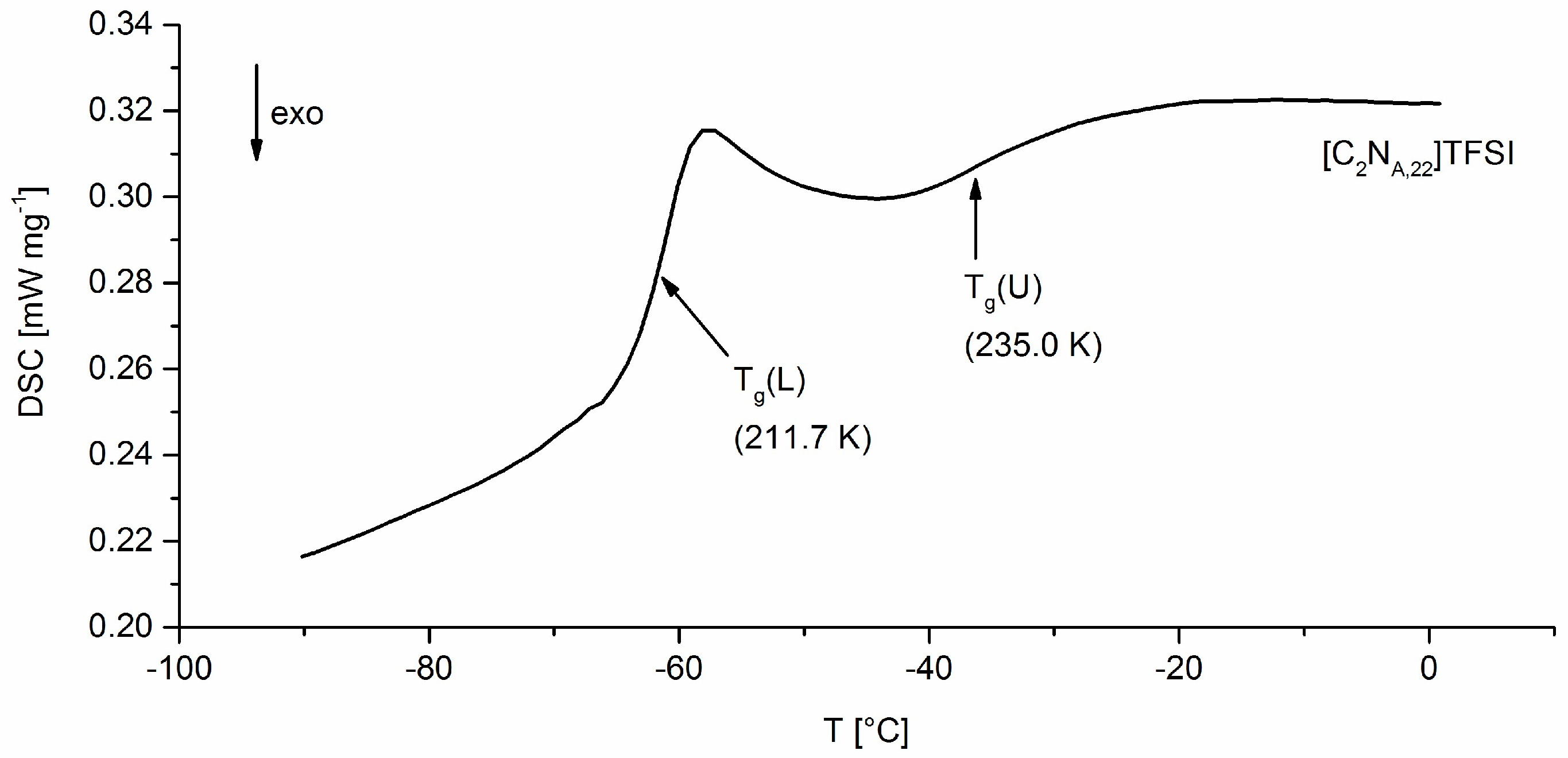
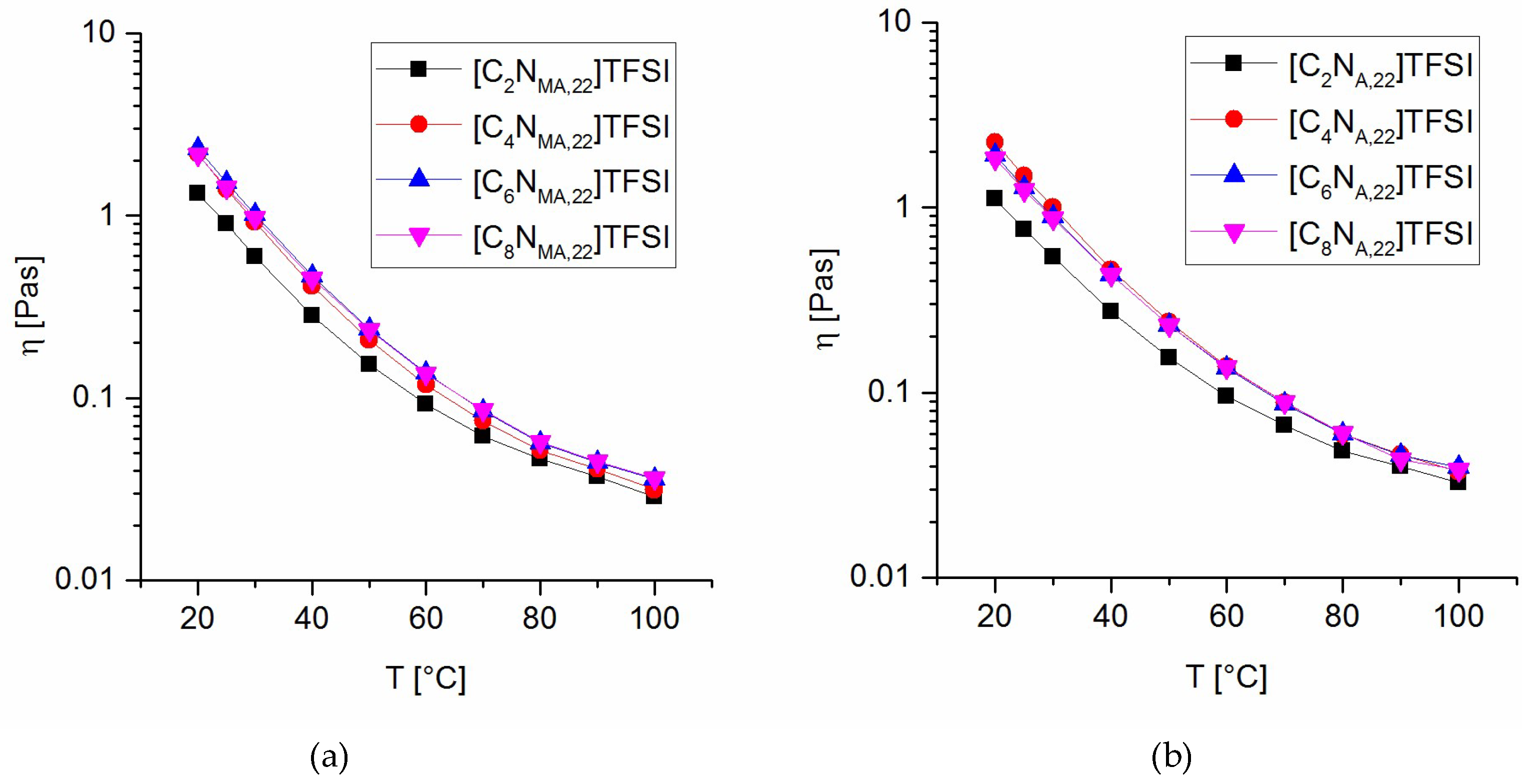
All solid products are stored in amber glass flasks inside an argon-filled glovebox. All TFSI ionic liquids are diluted to 50 wt% in acetone under argon and stored under exclusion of light at −30 °C. After removing the acetone, the ILs show less than 10 ppm water in Karl Fischer-titration. For synthesis and washing of ionic liquids deionized water (HPLC grade) was used without exceptions.
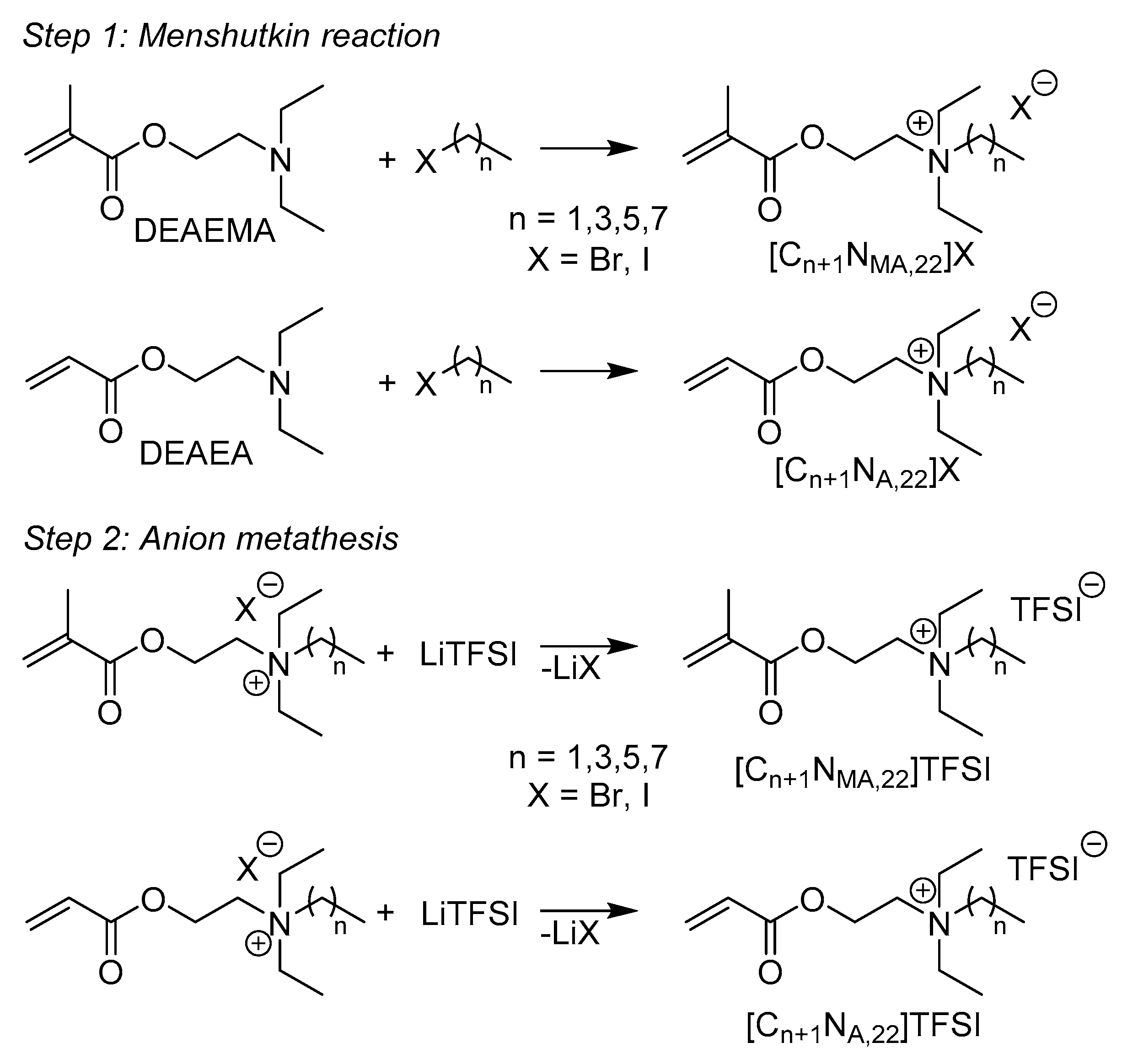
N,N,N-Triethyl-N-[2-(methacryloyloxy)ethyl]ammonium bromide ([C2NMA,22]Br)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 10.99 g, 59.32 mmol) and a excess of bromoethane (9.24 g, 84.84 mmol) in 30 mL acetone was stirred under argon-oxygen atmosphere (80:20% v/v) at 47 °C for 6 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum for 10 h. The product was received as a white powder (10.61 g, 60.8% yield). M.p.: 111.2 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 1.45 (t, 9H, CH3); 1.95 (s, 3H, CH3); 3.64 (q, 6H, CH2); 3.97 (t, 2H, CH2);); 4.76 (t, 2H, CH2); 5.68 (s, 1H, CH2); 6.12 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 8.3 (CH3); 18.3 (CH2); 54.4 (CH2); 56.0 (CH2); 57.8 (CH2); 127.5 (CH2); 135.1 (C); 166.4 (C=O); IR (cm−1): 2979 ν(C-H); 1717 ν(C=O); 1638 ν(C=C); 1479 δas(CH2); 1390 δs(CH3); 1320 δ(CH2); 1298 ν(C-O); 1168 ν(C-O); 1002 δ(CH2); 947 δ(C-H); 784 δ(CH2);
N-Butyl-N,N-diethyl-N-[2-(methacryloyloxy)ethyl]ammonium bromide ([C4NMA,22]Br)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 11.12 g, 60.00 mmol) and a excess of 1-bromobutane (11.51 g, 84.00 mmol) in 30 mL acetone was stirred under argon-oxygen atmosphere (80:20% v/v) under reflux for 11 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum for 24 h. The product was received as a white powder (9.49 g, 49.1% yield). M.p.:107.9 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 1.02 (t, 3H, CH3); 1.47 (m, 8H, CH3, CH2); 1.76 (m, 2H, CH2); 1.96 (s, 3H, CH3); 3.44 (m, 2H, CH2); 3.67 (q, 4H, CH2); 4.01 (t, 2H, CH2); 4.69 (t, 2H, CH2); 5.70 (s, 1H, CH2); 6.14 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 8.4 (CH3); 13.7 (CH3); 18.3 (CH3); 19.8 (CH2); 24.1 (CH2); 55.4 (CH2); 56.6 (CH2); 57.8 (CH2); 58.9 (CH2); 127.5 (CH2); 135.1 (C); 166.5 (C=O); IR (cm−1): 2963 ν(C-H); 1721 ν(C=O); 1640 ν(C=C); 1457 δas(CH2); 1399 δs(CH3); 1300 ν(C-O); 1159 ν(C-O); 1013 δ(CH2); 934 δ(C-H); 788 δ(CH2); Elemental analysis calcd (%) for C14H28BrNO2: C, 52.18; H, 8.76; N, 4.35. Found (%): C, 51.59; H, 8.49; N, 4.41.
N,N-Diethyl-N-hexyl-N-[2-(methacryloyloxy)ethyl]ammonium bromide ([C6NMA,22]Br)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 5.59 g, 30.02 mmol) and a excess of 1-bromohexane (6.93 g, 42.00 mmol) in 15 mL acetonitrile was stirred under argon-oxygen atmosphere (80:20% v/v) at 56 °C for 6 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (5.67 g, 53.9% yield). M.p.:102.7 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 0.87 (t, 3H, CH3); 1.33 (m, 6H, CH2); 1.43 (t, 6H, CH3); 1.74 (m, 2H, CH2); 1.93 (s, 3H, CH3); 3.40 (m, 2H, CH2); 3.64 (q, 4H, CH2); 3.97 (t, 2H, CH2); 4.65 (t, 2H, CH2); 5.66 (s, 1H, CH2); 6.11 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 8.3 (CH3); 16.9 (CH3); 18.3 (CH3); 22.2 (CH2); 22.4 (CH2); 26.1 (CH2); 31.2 (CH2); 55.0 (CH2); 56.6 (CH2); 57.9 (CH2); 59.0 (CH2); 127.4 (CH2); 135.1 (C); 166.4 (C=O); IR (cm−1): 2928 ν(C-H); 1717 ν(C=O); 1640 ν(C=C); 1446 δas(CH2); 1399 δs(CH3); 1298 ν(C-O); 1163 ν(C-O); 1009 δ(CH2); 945 δ(C-H); 813 δ(CH2); Elemental analysis calcd (%) for C16H32BrNO2: C, 54.85; H, 9.21; N, 4.00. Found (%): C, 54.73; H, 9.17; N, 4.01.
N,N,N-Triethyl-N-[2-(methacryloyloxy)ethyl]ammonium iodide ([C2NMA,22]I)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 27.789 g, 150.0 mmol), a slight excess of iodoethane (25.735 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 45 °C for 24 h and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (49.32 g, 96.4% yield). M.p.: 102.7 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.46 (t, 9H, CH3); 1.96 (s, 3H, CH3); 3.73 (q, 6H, CH2); 4.02 (t, 2H, CH2); 4.72 (t, 2H, CH2); 5.74 (s, 1H, CH2); 6.14 (s, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.6 (CH3); 17.6 (CH2); 53.9 (CH2); 55.6 (CH2); 58.0 (CH2); 126.1 (CH2); 135.8 (C); 166.1 (C=O); IR (cm−1): 2974 ν(C-H); 1719 ν(C=O); 1635 ν(C=C); 1457 δas(CH2); 1397 δs(CH3); 1300 ν(C-O); 1165 ν(C-O); 1000 δ(CH2); 949 δ(C-H); 782 δ(CH2); Elemental analysis calcd (%) for C12H24INO2: C, 42.24; H, 7.09; N, 4.10. Found (%): C, 42.24; H, 7.11; N, 4.10.
N-Butyl-N,N-diethyl-N-[2-(methacryloyloxy)ethyl]ammonium iodide ([C4NMA,22]I)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 27.789 g, 150.0 mmol), a slight excess of 1-iodobutane (30.363 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 60 °C for 3 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a slightly yellowish powder (54.02 g, 97.5% yield). M.p.: 116.1 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.02 (t, 3H, CH3); 1.49 (m, 8H, CH3, CH2); 1.90 (m, 2H, CH2); 1.96 (t, 3H, CH3); 3.62 (m, 2H, CH2); 3.75 (q, 4H, CH2); 4.00 (m, 2H, CH2); 4.72 (m, 2H, CH2); 5.75 (t, 1H, CH2); 6.15 (s, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.4 (CH3); 13.0 (CH3); 17.5 (CH3); 19.5 (CH2); 23.7 (CH2); 54.4 (CH2); 56.0 (CH2); 57.9 (CH2); 58.2 (CH2); 126.0 (CH2); 135.8 (C); 166.0 (C=O); IR (cm−1): 2963 ν(C-H); 1724 ν(C=O); 1640 ν(C=C); 1458 δas(CH2); 1401 δs(CH3); 1300 ν(C-O); 1163 ν(C-O); 1013 δ(CH2); 960 δ(C-H); 813 δ(CH2); Elemental analysis calcd (%) for C14H28INO2: C, 45.53; H, 7.64; N, 3.79. Found (%): C, 45.48; H, 7.64; N, 3.73.
N,N-Diethyl-N-hexyl-N-[2-(methacryloyloxy)ethyl]ammonium iodide ([C6NMA,22]I)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 27.789 g, 150.0 mmol), a slight excess of 1-iodohexane (34.992 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 60 °C for 3 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (57.18 g, 95.9% yield). M.p.: 95.7 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 0.91 (t, 3H, CH3); 1.39 (m, 6H, CH2); 1.49 (t, 6H, CH3); 1.92 (m, 2H, CH2); 2.00 (s, 3H, CH3); 3.62 (m, 2H, CH2); 3.75 (q, 4H, CH2); 4.01 (m, 2H, CH2); 4.72 (s, 2H, CH2); 5.75 (s, 1H, CH2); 6.15 (s, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.4 (CH3); 13.3 (CH3); 17.5 (CH3); 21.7 (CH2); 22.2 (CH2); 25.8 (CH2); 31.1 (CH2); 54.4 (CH2); 56.0 (CH2); 57.9 (CH2); 58.4 (CH2); 126.0 (CH2); 135.8 (C); 166.0 (C=O); IR (cm−1): 2954 ν(C-H); 1721 ν(C=O); 1640 ν(C=C); 1456 δas(CH2); 1403 δs(CH3); 1296 ν(C-O); 1157 ν(C-O); 1060 δ(CH2); 1011 δ(CH2); 936 δ(C-H); 808 δ(CH2); Elemental analysis calcd (%) for C16H32INO2: C, 48.37; H, 8.12; N, 3.53. Found (%): C, 48.43; H, 8.13; N, 3.50.
N,N-Diethyl-N-[2-(methacryloyloxy)ethyl]-N-octylammonium iodide ([C8NMA,22]I)
A mixture of 2-(diethylamino)ethyl methacrylate (DEAEMA, 27.789 g, 150.0 mmol), a slight excess of 1-iodooctane (39.621 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 75 °C for 3 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (59.47 g, 93.2% yield). M.p.: 80.5 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 0.88 (m, 3H, CH3); 1.32 (m, 6H, CH2); 1.46 (m, 10H, CH3, CH2); 1.91 (m, 2H, CH2); 1.96 (s, 3H, CH3); 3.64 (m, 2H, CH2); 3.76 (q, 4H, CH2); 4.06 (s, 2H, CH2); 4.73 (s, 2H, CH2); 5.74 (s, 1H, CH2); 6.15 (s, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.9 (CH3); 13.6 (CH3); 17.7 (CH3); 22.0 (CH2); 22.4 (CH2); 26.2 (CH2); 29.0 (CH2); 31.6 (CH2); 54.6 (CH2); 56.3 (CH2); 58.2 (CH2); 58.6 (CH2); 126.1 (CH2); 135.9 (C); 166.1 (C=O); IR (cm−1): 2928 ν(C-H); 1724 ν(C=O); 1640 ν(C=C); 1454 δas(CH2); 1399 δs(CH3); 1291 ν(C-O); 1154 ν(C-O); 1060 δ(CH2); 1009 δ(CH2); 936 δ(C-H); 806 δ(CH2); Elemental analysis calcd (%) for C18H36INO2: C, 50.82; H, 8.53; N, 3.29. Found (%): C, 50.51; H, 8.48; N, 3.27.
N-[2-(Acryloyloxy)ethyl]-N,N,N-triethylammonium iodide ([C2NA,22]I)
A mixture of 2-(diethylamino)ethyl acrylate (DEAEA, 25.686 g, 150.0 mmol), a slight excess of iodoethane (25.735 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 45 °C for 24 h and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (48.33 g, 98.5% yield). M.p.: 93.1 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.47 (t, 9H, CH3); 3.72 (q, 6H, CH2); 3.97 (m, 2H, CH2); 4.73 (s, 2H, CH2); 6.01 (dd, 1H, CH2); 6.24 (m, 1H, CH); 6.44 (dd, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.3 (CH3); 53.8 (CH2); 55.4 (CH2); 57.5 (CH2); 127.8 (CH); 131.6 (CH2); 164.8 (C=O); IR (cm−1): 2979 ν(C-H); 1719 ν(C=O); 1624 ν(C=C); 1461 δas(CH2); 1401 δs(CH3); 1271 ν(C-O); 1183 ν(C-O); 1086 δ(CH2); 1064 δ(CH2); 1020 δ(CH2); 998 δ(C-H); 960 δ(C-H); 808 δ(CH2); Elemental analysis calcd (%) for C11H22INO2: C, 40.38; H, 6.78; N, 4.28. Found (%): C, 40.61; H, 6.78; N, 4.52.
N-[2-(Acryloyloxy)ethyl]-N-butyl-N,N-diethylammonium iodide ([C4NA,22]I)
A mixture of 2-(diethylamino)ethyl acrylate (DEAEA, 15.945 g, 93.1 mmol), a slight excess of 1-iodobutane (18.849 g, 102.4 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 75 °C for 2 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a slightly yellowish powder (31.96 g, 96.6% yield). M.p.: 84.0 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.02 (t, 3H, CH3); 1.47 (m, 8H, CH3, CH2); 1.89 (m, 2H, CH2); 3.63 (m, 2H, CH2); 3.75 (q, 4H, CH2); 4.01 (m, 2H, CH2); 4.74 (s, 2H, CH2); 6.02 (m, 1H, CH2); 6.24 (m, 1H, CH); 6.44 (m, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.6 (CH3); 13.1 (CH2); 19.5 (CH2); 23.8 (CH2); 54.5 (CH2); 56.1 (CH2); 57.7 (CH2); 58.3 (CH2); 127.8 (CH); 131.7 (CH2); 164.8 (C=O); IR (cm−1): 2965 ν(C-H); 1724 ν(C=O); 1622 ν(C=C); 1459 δas(CH2); 1406 δs(CH3); 1260 ν(C-O); 1188 ν(C-O); 1071 δ(CH2); 1022 δ(CH2); 965 δ(C-H); 806 δ(CH2); Elemental analysis calcd (%) for C13H26INO2: C, 43.95; H, 7.38; N, 3.94. Found (%): C, 44.18; H, 7.38; N, 4.00.
N-[2-(Acryloyloxy)ethyl]-N,N-diethyl-N-hexylammonium iodide ([C6NA,22]I)
A mixture of 2-(diethylamino)ethyl acrylate (DEAEA, 25.686 g, 150.0 mmol), a slight excess of 1-iodohexane (34.992 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 60 °C for 3 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (55.26 g, 96.1% yield). M.p.: 81.1 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 0.91 (t, 3H, CH3); 1.41 (m, 6H, CH2); 1.48 (t, 6H, CH3); 1.92 (m, 2H, CH2); 3.62 (q, 2H, CH2); 3.74 (s, 4H, CH2); 3.99 (m, 2H, CH2); 4.74 (s, 2H, CH2); 6.02 (m, 1H, CH2); 6.24 (m, 1H, CH); 6.45 (m, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.4 (CH3); 13.3 (CH2); 21.8 (CH2); 22.2 (CH2); 25.8 (CH2); 31.1 (CH2); 54.4 (CH2); 56.0 (CH2); 57.6 (CH2); 58.5 (CH2); 127.8 (CH); 131.6 (CH2); 164.8 (C=O); IR (cm−1): 2954 ν(C-H); 1721 ν(C=O); 1624 ν(C=C); 1456 δ(CH2); 1406 δs(CH3); 1263 ν(C-O); 1188 ν(C-O); 1071 δ(CH2); 1022 δ(CH2); 971 δ(C-H); 936 δ(C-H); 806 δ(CH2); Elemental analysis calcd (%) for C15H30INO2: C, 47.00; H, 7.89; N, 3.65. Found (%): C, 47.05; H, 7.89; N, 3.66.
N-[2-(Acryloyloxy)ethyl]-N,N-diethyl-N-octylammonium iodide ([C8NA,22]I)
A mixture of 2-(diethylamino)ethyl acrylate (DEAEA, 25.686 g, 150.0 mmol), a slight excess of 1-iodooctane (39.621 g, 165.0 mmol) and phenothiazine (0.6 g, 3.0 mmol) as inhibitor in 15 mL acetonitrile was stirred under argon atmosphere at 60 °C for 3 days and under exclusion of UV irradiation. All volatile components were removed under reduced pressure. The precipitate was washed with ethyl ether and dried under vacuum. The product was received as a white powder (55.16 g, 89.4% yield). M.p.: 54.4 °C; 1H NMR (500 MHz, acetone-D6): δ (ppm) 0.89 (t, 3H, CH3); 1.33 (m, 6H, CH2); 1.47 (m, 10H, CH3, CH2); 1.92 (m, 2H, CH2); 3.62 (m, 2H, CH2); 3.75 (q, 4H, CH2); 4.01 (m, 2H, CH2); 4.74 (s, 2H, CH2); 6.01 (m, 1H, CH2); 6.24 (m, 1H, CH); 6.45 (m, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.5 (CH3); 13.5 (CH2); 21.9 (CH2); 22.4 (CH2); 26.2 (CH2); 28.9 (CH2); 31.6 (CH2); 54.5 (CH2); 56.1 (CH2); 57.7 (CH2); 58.5 (CH2); 127.8 (CH); 131.6 (CH2); 164.8 (C=O); IR (cm−1): 2926 ν(C-H); 1728 ν(C=O); 1640 ν(C=C); 1454 δas(CH2); 1406 δs(CH3); 1267 ν(C-O); 1188 ν(C-O); 1075 δ(CH2); 1009 δ(CH2); 978 δ(C-H); 804 δ(CH2); Elemental analysis calcd (%) for C17H34INO2: C, 49.64; H, 8.33; N, 4.40. Found (%): C, 49.46; H, 8.22; N, 3.35.
N,N,N-Triethyl-N-[2-(methacryloyloxy)ethyl]ammonium bis(trifluoromethanesulfonyl)imide ([C2NMA,22]TFSI)
The respective intermediate [C2NMA,22]I (22.180 g, 65.0 mmol) was dissolved in water (15 mL) and added to a solution of LiTFSI (19.501 g, 67.9 mmol) in water (15 mL). The ionic liquid precipitated instantly and the mixture was stirred further for 10 min at room temperature. The aqueous top layer was removed and the remaining organic layer was washed six times with water (40 mL), respectively. No iodide ions were detected in the last washing water by AgNO3 solution. The ionic liquid was diluted in acetone (40 mL) and dried over molecular sieves (10 g, 3 Å) for 24 h. The solution was filtrated (PTFE, 0.2 µm) and the acetone removed under reduced pressure. The ionic liquid was stirred at room temperature under vacuum (6 mbar) for 2 days to give the pure product as colorless oil (29.16 g, 90.7%yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 1.28 (t, 9H, CH3); 1.87 (s, 3H, CH3); 3.32 (q, 6H, CH2); 3.53 (m, 2H, CH2); 4.45 (m, 2H, CH2); 5.62 (m, 1H, CH2); 6.03 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.3 (CH3); 18.0 (CH2); 54.0 (CH2); 55.3 (CH2); 57.3 (CH2); 120.1 (q, CF3); 127.3 (CH2); 135.1 (C); 166.4 (C=O); IR (cm−1): 2996 ν(C-H); 1726 ν(C=O); 1638 ν(C=C); 1457 δas(CH2); 1399 δs(CH3); 1349 ν(SO2); 1179 ν(C-O); 1135 ν(S=O); 1053 ν(S=O); 1002 δ(CH2); 742 δs (CF3); 654 δ(SNS); 614 δa(SO2); Elemental analysis calcd (%) for C14H24F6N2O6S2: C, 34.01; H, 4.89; N, 5.67. Found (%): C, 34.11; H, 4.80; N, 5.73.
One-pot synthesis of [C2NMA,22]TFSI
DEAEMA (3.149 g, 17.0 mmol) and 1-iodoethane (2.917 g, 18.7 mmol) were added to a solution of LiTFSI (5.100 g, 17.8 mmol) in acetonitrile (5 mL). The mixture was stirred at elevated temperature (45 °C) for 16 h. All volatile components were removed under reduced pressure. The remaining oily phase was washed with water (120 mL in total) in six portions. The IL phase was dissolved in acetone (15 mL) and the solution was dried over molecular sieves (2.5 g, 3 Å) for 3 days. The solution was filtrated (PTFE, 0.2 µm) and the acetone was removed under reduced pressure. The ionic liquid was stirred at room temperature under vacuum (6 mbar) to give the pure product as colorless oil (5.56 g, 66.2% yield).
N-Butyl-N,N-diethyl-N-[2-(methacryloyloxy)ethyl]ammonium bis(trifluoromethanesulfonyl)imide ([C4NMA,22]TFSI)
The respective intermediate [C4NMA,22]I (24.003 g, 65.0 mmol) was suspended in water (30 mL) and the supernatant water was added to a solution of LiTFSI (19.501 g, 67.9 mmol) in water (15 mL). The undissolved intermediate salt was solved in acetone (15 mL) and added dropwise to the aqueous phase. The ionic liquid precipitated instantly and the mixture was stirred for 10 min at room temperature. Further processing was realized as described above for [C2NMA,22]TFSI to give the product as colorless oil (30.17 g, 88.8% yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 0.94 (t, 3H, CH3); 1.30 (t, 6H, CH3); 1.36 (q, 2H, CH2); 1.62 (m, 2H, CH2); 1.90 (s, 3H, CH3); 3.18 (m, 2H, CH2); 3.35 (q, 4H, CH2); 3.57 (m, 2H, CH2); 4.47 (m, 2H, CH2); 5.64 (s, 1H, CH2); 6.06 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.4 (CH3); 13.3 (CH3); 18.0 (CH3); 19.4 (CH2); 23.5 (CH2); 54.5 (CH2); 55.8 (CH2); 57.2 (CH2); 58.4 (CH2); 119.8 (q, CF3); 127.2 (CH2); 135.0 (C); 166.4 (C=O); IR (cm−1): 2970 ν(C-H); 1724 ν(C=O); 1638 ν(C=C); 1459 δas(CH2); 1349 ν(SO2); 1181 ν(C-O); 1135 ν(S=O); 1053 ν(S=O); 949 δ(C-H); 788 δ(CH2); 742 δs (CF3); 614 δ(SNS); Elemental analysis calcd (%) for C16H28F6N2O6S2: C, 36.78; H, 5.40; N, 5.36. Found (%): C, 37.10; H, 5.26; N, 5.37.
N,N-Diethyl-N-hexyl-N-[2-(methacryloyloxy)ethyl]ammonium bis(trifluoromethanesulfonyl)imide ([C6NMA,22]TFSI)
The respective intermediate [C6NMA,22]I (26.74 g, 65.0 mmol) was dissolved in acetone (40 mL) and added dropwise to a solution of LiTFSI (19.501 g, 67.9 mmol) in water (40 mL). The ionic liquid precipitated instantly and the mixture was stirred for 10 min at room temperature. Further processing was realized as described above for [C2NMA,22]TFSI to give the product as colorless oil (32.81 g, 91.7% yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 0.85 (t, 3H, CH3); 1.30 (m, 12H, CH3, CH2); 1.63 (m, 2H, CH2); 1.90 (s, 3H, CH3); 3.17 (m, 2H, CH2); 3.36 (q, 4H, CH2); 3.57 (m, 2H, CH2); 4.47 (s, 2H, CH2); 5.64 (s, 1H, CH2); 6.06 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.3 (CH3); 13.7 (CH3); 18.0 (CH3); 21.6 (CH2); 22.3 (CH2); 25.7 (CH2); 30.9 (CH2); 54.4 (CH2); 55.8 (CH2); 57.2 (CH2); 58.6 (CH2); 119.8 (q, CF3); 127.2 (CH2); 135.0 (C); 166.3 (C=O); IR (cm−1): 2963 ν(C-H); 2932 ν(C-H); 2864 ν(C-H); 1724 ν(C=O); 1638 ν(C=C); 1459 δas(CH2); 1399 δs(CH3); 1348 ν(SO2); 1181 ν(C-O); 1135 ν(S=O); 1055 ν(S=O); 949 δ(C-H); 788 δ(CH2); 742 δs (CF3); 654 δ(SNS); 616 δa(SO2); Elemental analysis calcd (%) for C18H32F6N2O6S2: C, 39.27; H, 5.86; N, 5.09. Found (%): C, 39.48; H, 5.74; N, 5.07.
N,N-Diethyl-N-[2-(methacryloyloxy)ethyl]-N-octylammonium bis(trifluoromethanesulfonyl)imide ([C8NMA,22]TFSI)
The IL was prepared accordingly to the procedure described above for [C6NMA,22]TFSI starting from [C8NMA,22]I (27.65 g, 65.0 mmol) in acetone (40 mL) and LiTFSI (19.501 g, 67.9 mmol) in water (40 mL). The product was received as colorless oil (34.11 g, 90.7% yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 0.87 (t, 3H, CH3); 1.27 (m, 6H, CH3); 1.33 (m, 10H, CH2); 1.65 (s, 2H, CH2); 1.93 (s, 3H, CH3); 3.20 (m, 2H, CH2); 3.38 (q, 4H, CH2); 3.59 (m, 2H, CH2); 4.50 (m, 2H, CH2); 5.67 (s, 1H, CH2); 6.09 (s, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.4 (CH3); 14.0 (CH3); 18.0 (CH3); 21.7 (CH2); 22.5 (CH2); 26.1 (CH2); 28.9 (CH2); 31.5 (CH2); 54.5 (CH2); 55.8 (CH2); 57.2 (CH2); 58.6 (CH2); 119.8 (q, CF3); 127.2 (CH2); 135.0 (C); 166.4 (C=O); IR (cm−1): 2959 ν(C-H); 2932 ν(C-H); 2857 ν(C-H); 1726 ν(C=O); 1638 ν(C=C); 1461 δas(CH2); 1399 δs(CH3); 1351 ν(SO2); 1183 ν(C-O); 1135 ν(S=O); 1055 ν(S=O); 947 δ(C-H); 788 δ(CH2); 742 δs (CF3); 654 δ(SNS); 616 δa(SO2); Elemental analysis calcd (%) for C20H36F6N2O6S2: C, 41.52; H, 6.27; N, 4.84. Found (%): C, 41.64; H, 6.05; N, 4.78.
N-[2-(Acryloyloxy)ethyl]-N,N,N-triethylammonium bis(trifluoromethanesulfonyl)imide ([C2NA,22]TFSI)
The IL was prepared accordingly to the procedure described above for [C2NMA,22]TFSI starting from [C2NA,22]I (24.451 g, 75.0 mmol) in water (15 mL) and LiTFSI (22.501 g, 68.38 mmol) in water (15 mL). The product was received as colorless oil (30.64 g, 85.0% yield). 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.44 (t, 9H, CH3); 3.62 (q, 6H, CH2); 3.83 (m, 2H, CH2); 4.67 (s, 2H, CH2); 6.00 (dd, 1H, CH2); 6.22 (m, 1H, CH); 6.42 (dd, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 6.9 (CH3); 53.7 (CH2); 55.1 (CH2); 57.3 (CH2); 120.1 (q, CF3); 127.6 (CH); 131.7 (CH2); 164.8 (C=O); IR (cm−1): 2996 ν(C-H); 1730 ν(C=O); 1638 ν(C=C); 1461 δas(CH2); 1410 δs(CH3); 1349 ν(SO2); 1177 ν(C-O); 1135 ν(S=O); 1053 ν(S=O); 996 δ(CH2); 788 δ(CH2); 742 δs (CF3); 654 δ(SNS); 614 δa(SO2); Elemental analysis calcd (%) for C13H22F6N2O6S2: C, 32.50; H, 4.62; N, 5.83. Found (%): C, 32.45; H, 4.40; N, 5.83.
N-[2-(Acryloyloxy)ethyl]-N-butyl-N,N-diethylammonium bis(trifluoromethanesulfonyl)imide ([C4NA,22]TFSI)
The IL was prepared accordingly to the procedure described above for [C2NMA,22]TFSI starting from [C4NA,22]I (26.645 g, 75.0 mmol) in water (20 mL) and LiTFSI (22.501 g, 68.38 mmol) in water (15 mL). The product was received as colorless oil (30.49 g, 80.5% yield). 1H NMR (500 MHz, acetone-D6): δ (ppm) 1.00 (s, 3H, CH3); 1.43 (s, 8H, CH3, CH2); 1.83 (s, 2H, CH2); 3.48 (s, 2H, CH2); 3.62 (s, 4H, CH2); 3.84 (s, 2H, CH2); 4.67 (s, 2H, CH2); 6.00 (m, 1H, CH2); 6.21 (m, 1H, CH); 6.42 (m, 1H, CH2); 13C NMR (125 MHz, acetone-D6): δ (ppm) 7.0 (CH3); 12.9 (CH2); 19.4 (CH2); 23.4 (CH2); 54.2 (CH2); 55.7 (CH2); 57.3 (CH2); 58.1 (CH2); 120.1 (q, CF3); 127.6 (CH); 131.7 (CH2); 164.8 (C=O); IR (cm−1): 2961 ν(C-H); 2935 ν(C-H); 2866 ν(C-H); 1730 ν(C=O); 1638 ν(C=C); 1463 δas(CH2); 1408 δs(CH3); 1351 ν(SO2); 1179 ν(C-O); 1135 ν(S=O); 1055 ν(S=O); 790 δ(CH2); 742 δs (CF3); 654 δ(SNS); 616 δa(SO2); Elemental analysis calcd (%) for C15H26F6N2O6S2: C, 35.43; H, 5.15; N, 5.51. Found (%): C, 35.45; H, 5.15; N, 5.57.
N-[2-(Acryloyloxy)ethyl]-N,N-diethyl-N-hexylammonium bis(trifluoromethanesulfonyl)imide ([C6NA,22]TFSI)
The IL was prepared accordingly to the procedure described above for [C6NMA,22]TFSI starting from [C6NA,22]I (24.915 g, 65.0 mmol) in acetone (40 mL) and LiTFSI (19.501 g, 67.9 mmol) in water (30 mL). The product was received as colorless oil (31.20 g, 89.5% yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 0.89 (s, 3H, CH3); 1.33 (s, 12H, CH3, CH2); 1.66 (s, 2H, CH2); 3.21 (m, 2H, CH2); 3.38 (m, 4H, CH2); 3.60 (s, 2H, CH2); 4.52 (s, 2H, CH2); 5.96 (m, 1H, CH2); 6.11 (m, 1H, CH); 6.44 (m, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.4 (CH3); 13.7 (CH2); 21.7 (CH2); 22.3 (CH2); 25.8 (CH2); 31.0 (CH2); 54.5 (CH2); 55.8 (CH2); 56.9 (CH2); 58.7 (CH2); 119.8 (q, CF3); 126.9 (CH); 132.8 (CH2); 165.1 (C=O); IR (cm−1): 2972 ν(C-H); 2883 ν(C-H); 1732 ν(C=O); 1637 ν(C=C); 1463 δas(CH2); 1408 δs(CH3); 1349 ν(SO2); 1179 ν(C-O); 1135 ν(S=O); 1053 ν(S=O); 790 δ(CH2); 740 δs (CF3); 653 δ(SNS); 614 δa(SO2); Elemental analysis calcd (%) for C17H30F6N2O6S2: C, 38.06; H, 5.64; N, 5.22. Found (%): C, 37.70; H, 5.63; N, 5.13.
N-[2-(Acryloyloxy)ethyl]-N,N-diethyl-N-octylammonium bis(trifluoromethanesulfonyl)imide ([C8NA,22]TFSI)
The IL was prepared accordingly to the procedure described above for [C6NMA,22]TFSI starting from [C8NA,22]I (26.739 g, 65.0 mmol) in acetone (40 mL) and LiTFSI (19.501 g, 67.9 mmol) in water (30 mL). The product was received as colorless oil (32.77 g, 89.3% yield). 1H NMR (500 MHz, CDCl3): δ (ppm) 0.88 (t, 3H, CH3); 1.28 (m, 6H, CH2); 1.34 (t, 10H, CH3, CH2); 1.66 (s, 2H, CH2); 3.20 (m, 2H, CH2); 3.39 (q, 4H, CH2); 3.60 (m, 2H, CH2); 4.52 (m, 2H, CH2); 5.96 (dd, 1H, CH2); 6.12 (m, 1H, CH); 6.45 (dd, 1H, CH2); 13C NMR (125 MHz, CDCl3): δ (ppm) 7.4 (CH3); 14.0 (CH2); 21.8 (CH2); 22.5 (CH2); 26.1 (CH2); 28.9 (CH2); 31.5 (CH2); 54.5 (CH2); 55.8 (CH2); 56.9 (CH2); 58.7 (CH2); 119.8 (q, CF3); 126.9 (CH); 132.9 (CH2); 165.1 (C=O); IR (cm−1): 2959 ν(C-H); 2932 ν(C-H); 2860 ν(C-H); 1732 ν(C=O); 1638 ν(C=C); 1465 δas(CH2); 1408 δs(CH3); 1349 ν(SO2); 1179 ν(C-O); 1135 ν(S=O); 1055 ν(S=O); 790 δ(CH2); 742 δs (CF3); 656 δ(SNS); 616 δa(SO2); Elemental analysis calcd (%) for C19H34F6N2O6S2: C, 40.42; H, 6.07; N, 4.96. Found (%): C, 41.04; H, 6.01; N, 5.06.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}