1. Introduction
Neuroinflammation is one part of the immune reaction to harmful stimulation in the central nervous system. The function of the neuroinflammatory response is to remove necrotic cells and tissues caused by pathogenic infections, disease, and other damage. Uncontrolled neuroinflammation, however, contributes to the progress of neurodegenerative diseases including Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease [
1]. Microglial cells, brain-specific macrophages that reside in the central nervous system (CNS), play a critical role in maintaining the homeostasis of the brain and the neuroinflammatory response [
2]. Microglia contribute to the maintenance of synaptic homeostasis by removing debris such as damaged neurons and pathogens from the central nervous system [
3].
Abnormal activation of microglia by inflammatory stimuli, such as LPS, leads to the secretion of neurotoxic molecules including NO, prostaglandin (PG) E
2, and inflammatory cytokines. Overexpression of these inflammatory factors causes damage to nerve cells and ultimately leads to neurodegenerative disease [
4]. Recently, a number of studies have aimed at identifying agents which control the activation of microglial cells, with the ultimate aim of preventing or treating brain diseases associated with chronic inflammation. Some of these studies have involved natural substances.
Microglial cells respond to LPS stimulation by secreting inflammatory factors such as NO, PGE
2, and the proinflammatory cytokines interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF)-α [
5]. Levels of NO and PGE
2 are closely related to the expression of the enzymes which synthesize them, iNOS and COX-2. COX-2 and iNOS are important for the progression of several inflammatory diseases, in which iNOS production is suppressed by HO-1, an important component of the immune system, resulting in a negative feedback loop [
6]. Increases in the amount of HO-1 in activated microglia reduce the levels of proinflammatory factors such as NO, TNF-α, IL-6, and IL-1β, and the production of HO-1 is regulated by a redox-dependent transcription factor, Nrf-2 [
7]. In the activation phase of the inflammatory reaction, free Nrf-2 is translocated into the nucleus, where it binds to the antioxidant response element, which in turn induces the production of HO-1 [
8]. Some recent studies show that HO-1 is important for immunomodulation and anti-inflammatory effects.
NF-κB is an important transcription factor involved in inflammatory processes and the regulation of inflammatory factors such as cytokines, chemokines, inducible enzymes, growth factors, and immune receptors [
9]. NF-κB is potentially an important molecular target for the development of agents against inflammatory diseases, and many recent studies have focused on the regulation of NF-κB. The activity of NF-κB is closely related to that of the MAPK signaling pathway [
10]. MAPK is an intracellular enzyme that activates inflammatory processes in response to stimuli such as LPS from the extracellular environment. MAPK includes an extracellular signal-regulated kinase (ERK), p38, and members of the c-Jun NH
2-terminal kinase (JNK) subfamily. It plays a role in the regulation of inflammatory molecules and immune-related cytotoxic factors that induce expression of inflammatory gene in microglia or macrophages [
11]. MAPK, along with NF-κB, is therefore an important potential target for the treatment of inflammatory diseases [
12].
AGR is the root of Angelica gigas Nakai (Umbelliferae), known as “Danggui” in Korea. AGR is cultivated as a medicinal plant over a wide area of Asia, but is mainly produced in Korea, China, and Japan. Plants of different origin are used in the three countries. Angelica gigas Nakai is the “Danggui” commonly used in Korea. AGR is listed in the ancient medicine text “Shennong’s Classic of Materia Medica,” and is widely used in traditional Asian medicine for improving blood circulation and hematopoiesis. Recent research has shown that AGR promotes blood flow in the coronary arteries and stimulates red blood cell generation. However, the effect of AGR on neuroinflammation mediated by microglial cells and its effects on NF-κB, MAPK, and Nrf-2 has not previously been studied.
In this study, we investigated the effect of an ethanol extract of AGR on the inflammatory response using LPS stimulation in brain microglia BV2 cells. We also investigated how the potency of the extracts correlated with the activation or inactivation of the NF-κB, MAPK, and Nrf-2 signaling pathways. We investigated the chemical constituents of AGR ethanol extracts using HPLC.
3. Discussion
Microglia are phagocytic cells located in the CNS which regulate the initial immune response in the brain. They secrete NO, inflammatory cytokines, reactive oxygen species, and superoxide generated by uncontrolled excessive inflammatory reactions, and contribute to the development of degenerative brain diseases such as dementia [
13]. Therefore, the modulation of inflammatory mediators produced from microglia activated by excessive inflammatory responses has been identified as a therapeutic target in conjunction with amyloid beta (Aβ) and tau protein in recent studies of dementia. LPS, which is an endotoxin produced by Gram-negative bacteria, is a major producer of inflammation and specifically activates TLR4 pathway in macrophages and microglia cells, leading to the overproduction of NO, PGE
2, and proinflammatory cytokines, including IL-6, IL-1β, TNF-α, macrophage inflammatory protein (MIP)-1, monocyte chemoattractant protein (MCP)-1, and IL-16 [
14,
15]. These inflammatory mediators act as cytotoxic substances, which accelerate the inflammatory response and contribute to the initiation and maintenance of chronic inflammatory diseases [
15]. Therefore, effective control of the LPS-induced secretion of various inflammatory mediators, activation of enzymes, expression of related genes, and activation of the TLR4 mechanism is very important for preventing and treating not only inflammatory diseases of the CNS but also degenerative brain diseases. In this study, we investigated the effect of AGR, which has been widely used in food and medicine from ancient times in East Asia, on the inflammatory responses induced by LPS in BV2 microglia, in order to confirm the antineuroinflammatory activity of AGR and to analyze its mechanism.
Cultured BV2 cells were treated with AGR and incubated for 24 h to identify the cytotoxicity-free range of 100 μg/mL or less (
Figure 1A). The inhibitory effect of AGR on NO secretion was then tested at lower concentrations. NO is an active nitrogen species that is a toxic low-molecular radical involved in the defense functions of the immune system [
16]. NO is produced from L-arginine through constitutive nitric oxide synthase (cNOS) and iNOS. Excessive secretion of NO in the body causes cytotoxicity and mediates inflammatory responses in the CNS, leading to the onset of degenerative brain diseases [
17]. Proinflammatory cytokines play a diverse role in immune, infectious, and tissue repair processes, cell growth, and biological defenses, and are involved in the pathogenesis of many diseases that are directly or indirectly related to inflammation. BV2 microglia activated by LPS secrete cytokines including IL-6, IL-1β, TNF-α, and transforming growth factor (TGF)-β, which may act to promote inflammation. TNF-α and IL-1β are typical proinflammatory cytokines associated with degenerative diseases, and they induce inflammatory disease by affecting the synthesis of mediators such as PG, leukotriene, and NO [
18]. The levels of these cytokines are increased in the blood and brain tissue of patients with degenerative brain diseases, and they are known to play a pivotal role in the pathogenesis of neuronal apoptosis [
19]. According to the results of this study, pretreatment with AGR extract significantly decreased the amount of NO secreted from BV2 cells after LPS stimulation (
Figure 1B), due to the inhibition of mRNA and protein production of the iNOS protein (
Figure 2A,B). Some members of the PG family are known to exacerbate progressive inflammation and pain [
12]. PGE
2 and PGD
2, members of this family, are synthesized from arachidonic acid via the action of COX and lipoxygenase [
14]. COX-2, one of the COX isomers, is a major target of research into therapeutic agents such as nonsteroidal anti-inflammatory drugs, and considerable research has been conducted into COX-2-selective inhibitors. In this study, BV2 cells were shown to express COX-2 enzyme following LPS stimulation. Expression of the mRNA and protein was significantly reduced by pretreatment with AGR in a dose-dependent manner (
Figure 2A,B). This study examined the effects of AGR on the secretion of inflammatory cytokines and showed that pretreatment with AGR strongly inhibited the secretion of both TNF-α and IL-6 and the expression of their respective mRNAs (
Figure 1C–F). This effect was statistically significant and concentration-dependent at most treatment concentrations. Although the amount of IL-1β cytokine secretion in BV2 microglial cells was so low that it could not be detected by ELISA, mRNA expression, as analyzed by RT-qPCR, was strongly inhibited by AGR pretreatment as well as the expression of other cytokine genes (
Figure 1G).
HO decomposes heme to produce biliverdin, CO, and ferrous ions. Both biliverdin and bilirubin have antioxidative effects. CO in the air is a toxic gas, but HO-1-induced CO has anti-inflammatory properties [
20]. Therefore, the expression of HO-1 can represent antioxidative and anti-inflammatory action, which can affect the expression of iNOS induced by LPS in BV2 cells and may consequently inhibit NO secretion. Our results show that AGR extract effectively induces the production of HO-1 in BV2 microglia and has an inhibitory effect on the HO-1 induced production of NO and iNOS (
Figure 2C,D). Nrf-2 is a pivotal regulator of several antioxidant enzymes, such as HO-1, and catalyzes the degradation of heme to antioxidants such as biliverdin, CO, and iron [
21]. Nrf-2 liberated from cytoplasmic Kelch-like ECH-associated protein (Keap) 1 is transported into the nucleus and induces the production of HO-1, which plays an important role in inhibiting the inflammatory response. We also observed that the pattern of increase of Nrf-2 protein in the nucleus was similar to that of the production of HO-1 (
Figure 2C), which appears to directly affect the neuroinflammation inhibitory activity of AGR.
NF-κB is a transcription factor that plays a crucial role in the expression of genes that regulate cellular activities such as inflammation and cell differentiation [
22], is stimulated by TLR4 activation, and is separated from the inhibitor protein IκBα in the cytoplasm and translocated into the nucleus. NF-κB translocated into the nucleus binds to the promoter region of target genes and promotes the transcription of inflammatory substances such as inflammatory cytokines, adhesion molecules, and chemokines [
23]. In this study, we also analyzed the expression of NF-κB pathway proteins, in order to investigate the mechanisms by which extract of AGR inhibits neuroinflammation. It appears that AGR has an inhibitory effect on the production of inflammatory substances, in view of the rapid decrease of cytoplasmic NF-κB p65 protein by LPS stimulation, partial regeneration by AGR pretreatment, and reduction of p65 protein migration into the nucleus due to the regulation of NF-κB activation (
Figure 3A,B). Analysis of the expression of the inhibitory protein IκBα revealed that degradation and phosphorylation following LPS stimulation were inhibited by pretreatment with AGR, confirming that AGR has an inhibitory effect on NF-κB activation (
Figure 3C).
MAPKs are serine/threonine kinases that transport extracellular signals into the nucleus, and their signal transduction leads to the synthesis of mediators that induce inflammatory responses. Phosphorylation of MAPK induces the activation of transcription factors including NF-κB and activator protein (AP)-1, involved in the formation of free radicals such as NO, and playing an important role in the activation of early stage inflammatory responses [
24]. In BV2 microglia, phosphorylation of ERK, p38, and JNK MAPK by LPS was strongly suppressed by pretreatment with AGR, and showed high dependence on concentration (
Figure 4). These results suggest that one of the major causes of the neuroinflammatory inhibitory activity of AGR is its regulation of MAPK activity.
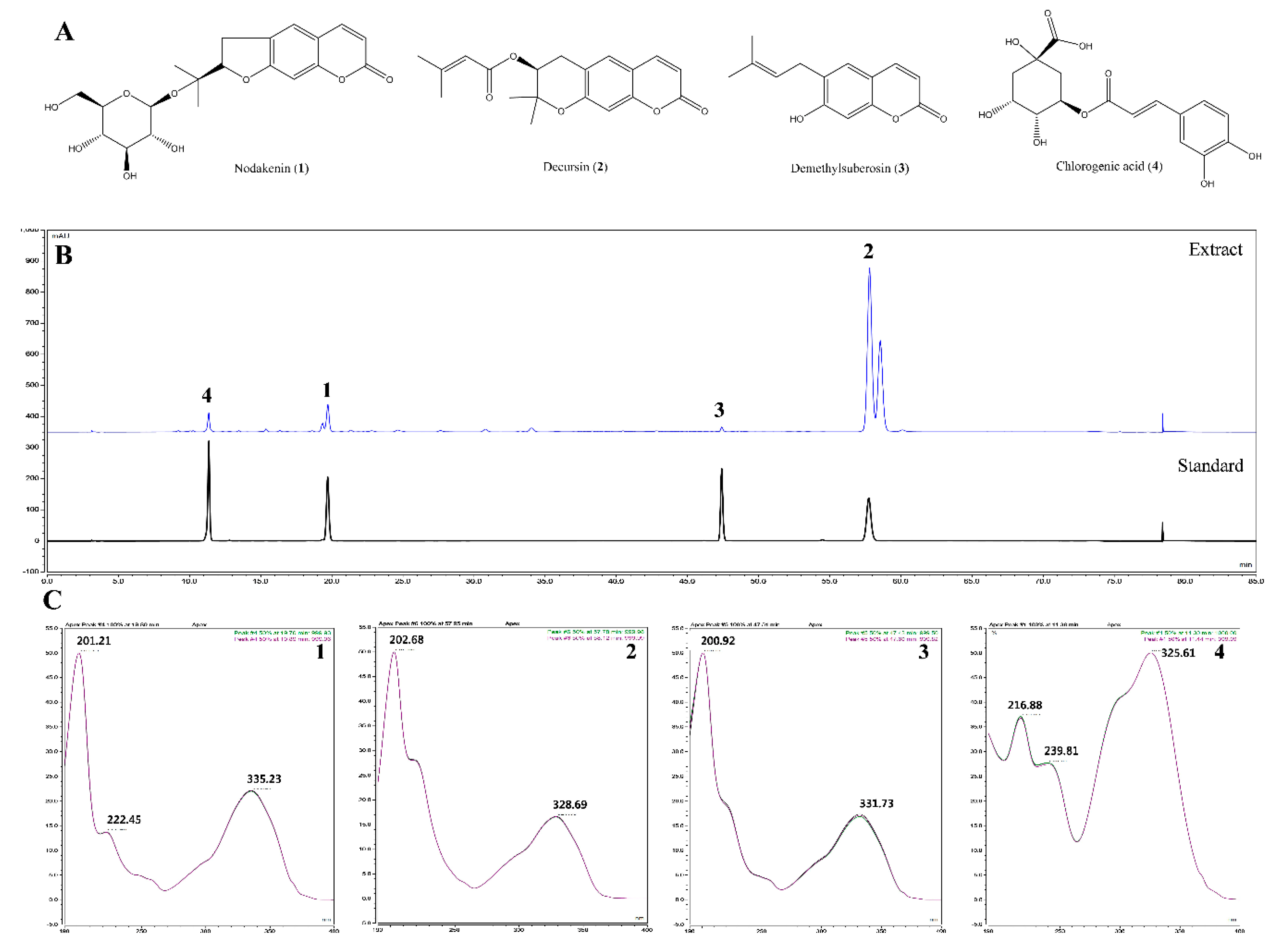
We also identified the main components (nodakenin, decursin, demethylsuberosin, and chlorogenic acid) of the AGR extract through HPLC analysis (
Figure 5). Previous studies of these components have shown that that nodakenin appears to have inhibitory effects on inflammation resulting from the regulation of TNF and NF-κB-related mechanisms in macrophages as observed by the inhibition of endotoxin shocks in mice [
25]. There have also been reports that decursin inhibits the activation of MAPK and NF-κB induced by Aβ in PC12 cells [
26]. Demethylsuberosin has also been reported to be effective in inhibiting the production of IL-6, NO and PGE
2 in mast cells and macrophages [
27,
28]. These studies indicate that the antineuroinflammatory efficacy of AGR is closely related to the activity of its three constituents, nodakenin, decursin, and demethylsuberosin. In order to confirm the antineuroinflammatory activity of these components, we evaluated the effect of nodakenin, decursin, and demethylsuberosin on the secretion of NO and inflammatory cytokines on the microglia and confirmed that the three compounds showed antineuroinflammatory activity (
Figure 6). In addition, the effects on the activation of NF-κB and MAPK were evaluated to explore the antineuroinflammatory mechanism of these components, and the results showed that the three components significantly inhibit the nuclear translocation of p65 protein, phosphorylation of IκBα, and phosphorylation of ERK, p38 MAPK (
Figure 7). The antineuroinflammatory activities and structural features observed for the three main components showed that the original coumarin (demethylsuberosin;
3) had stronger activity than furanocoumarin (nodakenin;
1) and pyranocoumarin (decursin;
2), which indicated that the coumarin skeleton plays an important role in anti-neuroinflammatory activity.
4. Materials and Methods
4.1. Materials and Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and antibiotics were purchased from HyClone (Logan, UT, USA). LPS from
Escherichia coli O55:B5, bovine serum albumin (BSA), and DEX were purchased from Sigma–Aldrich (St. Louis, MO, USA). Consumables used for cell culture, including dishes, well plates, and tubes, were purchased from Sarstedt (Nümbrecht, Germany). CCK and ELISA antibody sets were obtained from Dojindo Molecular Technologies (Kumamoto, Japan) and eBioscience (San Diego, CA, USA), respectively. Primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies for Western blotting were acquired from Cell Signaling Technology (Boston, MA, USA). Polyvinylidene fluoride (PVDF) membrane for Western blotting was obtained from Millipore (Bedford, MA, USA). RNA extraction kits were purchased from iNtRON Biotechnology (Daejeon, Korea). DNA synthesis kits, oligonucleotide primers, and AccuPower
® 2X Greenstar qPCR Master Mix (ROX) were purchased from Bioneer (Daejeon, Korea). AGR ethanol extract was obtained as a freeze-dried powder from the Korea Plant Extract Bank (Ochang, Korea), dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C before use. Chemical standards for AGR (nodakenin, decursin, demethylsuberosin, and chlorogenic acid) were purchased from ChemFaces (Wuhan, China). Information on the various reagents and antibodies used in this study is shown in
Table 2.
4.2. Microglial Cell Culture, Stimulation, and Treatment of Test Drugs
Mouse microglial BV2 cells were obtained from Prof. Kyoungho Suk at Kyungpook National University (Daegu, Korea). The BV2 cells were maintained in complete DMEM containing 1% antibiotic (100 U/mL penicillin and 100 μg/mL streptomycin) and 10% FBS. Cells were incubated in a humidified 5% CO2 atmosphere at 37 °C. All experiments were conducted 18 h after cell seeding in various culture plates. To stimulate the inflammatory responses of BV2 microglia, the culture medium was exchanged for fresh serum-free DMEM and 100 ng/mL of LPS was added in the presence or absence of AGR (10, 50, and 100 μg/mL) for the periods indicated.
4.3. Viability Assay for BV2 Microglia
To determine cell viability, BV2 microglia were plated in 96 well culture plates (5 × 104 cells/well) and were incubated with different concentrations of AGR and LPS for 48 h. The CCK solution was then treated and incubated for an additional hour. Cell viability was calculated from optical density by analyzing the color changes in each well using an ELISA reader at a wavelength of 450 nm using a SpectraMax i3 (Molecular Devices, San Jose, CA, USA).
4.4. Analysis of NO Secretion
Griess assays were performed to analyze the effect of AGR on the secretion of NO. Microglia cells were seeded in 96-well cell culture plates under the same conditions used for the viability test, and preincubated for 18 h. Three concentrations of ARG were pretreated and after 1 h 100 ng/mL LPS was added to the culture to induce an intracellular inflammatory response. After another 24 h, 100 μL of Griess reagent was added to each well of the 96-well plate. After incubation at room temperature for 5 min, absorbance was measured at 570 nm using an ELISA reader, in order to evaluate the levels of expression or inhibition of NO.
4.5. ELISA for the Analysis of Inflammatory Cytokine Secretion
ELISA experiments were conducted to investigate the effect of AGR extract on the secretion of the inflammatory cytokines TNF-α and IL-6 induced by LPS in BV2 cells. Each well of a 24-well cell culture plate was seeded with 2.5 × 105 cells. Preincubation and pretreatment of AGR and LPS treatments were performed under the same conditions as the previous Griess assay. After 6 h incubation, culture medium was retrieved, cell debris was removed by centrifugation, and the resulting medium used for ELISA analysis, performed according to the manufacturer’s standard protocol. The amount of each inflammatory cytokine was evaluated according to the intensity of its color after treatment with the TMB substrate, and the absorbance was read at a wavelength of 450 nm using an ELISA reader.
4.6. Preparation of Cytoplasmic and Nuclear Extracts for NF-κB and Nrf-2 Protein Detection
Cytoplasmic and nuclear extracts were separated according to the manufacturer’s protocol using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Rockford, IL, USA). Extracts were stored at −80 °C and processed using the same approach as the Western blotting of whole cell lysate.
4.7. Western Blotting for Protein Expression Measurement
To prepare samples for Western blotting, BV2 cells were seeded on a six-well plate at 1.5 × 10
6 cells per well and cultured overnight. After replacement with fresh serum-free medium, the AGR extract was treated at different concentrations and cultured for 1 h, and an inflammatory response was induced by LPS. Cultured cells were harvested at predetermined times and washed twice with ice-cold phosphate-buffered saline (PBS). The supernatant was collected following centrifugation, and the cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Millipore) and incubated on ice for 30 min. Following centrifugation at 15,000 rpm the supernatant was collected and normalized using Bradford’s method, followed by denaturation at 95 °C for 5 min. Protein samples were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes using transfer buffer. The non-specific binding sites of the membranes were blocked with 3% BSA for 1 h at room temperature, and then incubated overnight with specific primary antibodies at 4 °C. The membranes were then washed with tris-buffered saline (TBS)-T containing 0.1% Tween 20 nonionic detergent, incubated with HRP-conjugated secondary antibodies for 1 h at room temperature, and then analyzed using a Chemidoc system (BIO-RAD), with an enhanced chemiluminescence (ECL) solution. Each protein band was normalized according to β-actin expression and quantified using the ImageJ software (
https://imagej.nih.gov/ij/).
4.8. RNA Extraction and RT-qPCR
Total RNA was isolated using easy-BLUE
TM RNA (iNtRON Bio) extraction kits in accordance with the manufacturer’s protocol, and cDNA was synthesized from the purified RNA. A total of 1 μg of RNA was reverse transcribed into cDNA using AccuPower
® RT PreMix. The RT-qPCR oligonucleotide primers for mouse microglial cDNA are shown in
Table 3 [
29]. RT-qPCR reactions were performed in triplicate in 20 μL volumes with the following reagents: 0.3 μM final concentration of each primer with 1 μL each of forward and reverse primers, 10 μL of AccuPower
® 2× Greenstar qPCR master mix, 5 μL of template DNA, and 3 μL of RNase-free water. The following cycles were applied for the RT-qPCR of TNF-α, IL-6, IL-1β, iNOS, COX-2, HO-1, and β-actin: 40 cycles of 94 °C for 15 s and 60 °C for 1 min [
29]. Amplification and analysis were performed using the QuantStudio 6 Flex Real-time PCR System (Thermo Scientific), and samples were compared using the relative CT method. Fold changes in gene expression were determined relative to a blank control after normalization to β-actin expression using the 2
−ΔΔCT method.
4.9. Sample Preparation for HPLC Analysis
All sample solutions were dissolved in methanol. Standard stock solutions of compounds were prepared at a concentration of 1 mg/mL and were then serially diluted with methanol to obtain a calibration curve of standard solutions at various concentration levels ranging from 12.5–200 μg/mL for nodakenin (1) and chlorogenic acid (4); 37.5–600 μg/mL for decursin (2); and 3.125–50 μg/mL for demethylsuberosin (3). AGR extract was prepared at a concentration of 5 mg/mL. All solutions for analysis were filtered through 0.45 μm RC membrane syringe filters (Sartorius, Germany).
4.10. Optimization of Chromatographic Conditions
HPLC analysis was performed using a Dionex UltiMate 3000 system (Dionex Corp., Sunnyvale, CA, USA) equipped with a binary pump, autosampler, column oven, and DAD. System control and data analysis were carried out using Dionex Chromelon software. Separation was carried out on a Luna C18 column (250 × 4.6 mm, 5 μm, Phenomenex, Torrance, CA, USA), with the column oven temperature kept at 30 °C, at a UV wavelength of 330 nm. The mobile phase consisted of water (solvent A) and acetonitrile (solvent B) with gradient elution of 0–5 min, 5–15% B; 5–12 min, 15–20% B; 12–30 min, 20–25% B; 30–40 min, 25–50% B; 40–60 min, 50% B; and 60–70 min, 50–80% B, at a flow rate of 1.0 mL/min. Before injection of the next sample, the column was re-equilibrated with the initial gradient of solvents for 10 min.
4.11. Validation of the Method
Specificity was assessed by comparing the chromatographic profile of the standard with that of the sample extract. Linearity was assessed by computing the correlation coefficient (R2) of the calibration curve for each compound at concentrations ranging from 3.125 to 600 μg/mL. Regression equations were calculated using the equation y = ax ± b, where y and x are the peak areas and concentrations of the sample, respectively.
4.12. Preparation of the Main Components of AGR
The main components of ARG were identified using HPLC analysis, as described above. Each compound was dissolved in 100% DMSO. The stock concentration of the three compounds nodakenin, decursin, and demethylsuberosin was 50 mM, and the intracellular concentrations applied were 1, 10, 50, and 100 μM, (DMSO concentrations of 0.2% or less). The effects of these compounds on the viability of BV2 cells was measured, and their inhibitory effects on the secretion of NO and inflammatory cytokines induced by LPS was confirmed.
4.13. Statistical Analysis
Each result is an averaged value obtained from at least three independent experiments, and is expressed as mean ± standard error of the mean (SEM). Statistical significance was determined using one-way analysis of variance followed by Dunnett’s test, after comparing each treated group and the LPS group. Statistical significance was defined as * p < 0.05, ** p < 0.001, *** p < 0.0001 (vs. LPS).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}