
Reductive Metabolism of Ellagitannins in the Young Leaves of Castanopsis sieboldii

 ,
, 
Abstract
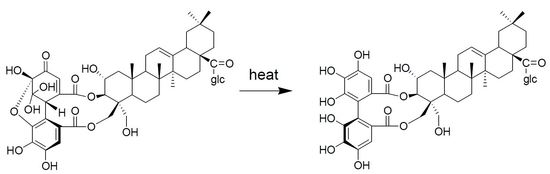
:
1. Introduction
2. Results and Discussion
2.1. HPLC Analysis of the Leaves
2.2. Structures of Phenazine Derivatives
2.3. Reaction of the Unstable Metabolites
2.4. Revision of Structures of Triterpene HHDP Esters
3. Materials and Methods
3.1. General Information
3.2. Plant Material
3.3. Extraction and HPLC Analysis
3.4. Isolation of Phenazine Derivatives
3.4.1. Compound 1o
3.4.2. Compound 1u
3.4.3. Compound 2o,u
3.5. Acid Hydrolysis of Compound 1o,u
3.6. Separation of Triterpene HHDP Esters from Mature Leaves
3.6.1. Castanopsinin E (3)
3.6.2. Castanopsinin C (4)
3.6.3. Castanopsinin A (9)
3.6.4. Castanopsinin B (10)
3.6.5. Castanopsinin F (11)
3.6.6. Castanopsinin H (12)
3.7. Alkaline Methanolysis of Triterpene HHDP Ester-Containing Fraction
3.8. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okuda, T.; Yoshida, T.; Hatano, T. Hydrolyzable tannins and related polyphenols. In Progress in the Chemistry of Organic Natural Products; Herz, W., Kirby, G.W., Moore, R.E., Steglich, W., Tamm, C., Eds.; Springer: New York, NY, USA, 1995; Volume 66, pp. 1–117. [Google Scholar]
- Haslam, E.; Cai, Y. Plant polyphenols (vegetable tannins): Gallic acid metabolism. Nat. Prod. Rep. 1994, 11, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hatano, T.; Ito, H.; Okuda, T. Structural diversity and antimicrobial activities of ellagitannins. In Chemistry and Biology of Ellagitannins, an Underestimated Class of Bioactive Plant Polyphenols; Quideau, S., Ed.; World Scientific Publishing: Singapore, 2009; pp. 55–93. [Google Scholar]
- Gross, G.G. Biosynthesis of ellagitannins: Old ideas and new solutions. In Chemistry and Biology of Ellagitannins, an Underestimated Class of Bioactive Plant Polyphenols; Quideau, S., Ed.; World Scientific Publishing: Singapore, 2009; pp. 94–118. [Google Scholar]
- Niemetz, R.; Gross, G.G. Enzymology of gallotannin and ellagitannin biosynthesis. Phytochemistry 2005, 66, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Niemetz, R.; Gross, G.G. Ellagitannin biosynthesis: Laccase-catalyzed dimerization of tellimagrandin II to cornusiin E in Tellima grandiflora. Phytochemistry 2003, 64, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, T.; Matsuo, Y.; Saito, Y.; Tanaka, T. Enzymatic oxidation of ellagitannin and a new ellagitannin metabolite from Camellia japonica leaves. Tetrahedron 2017, 73, 500–507. [Google Scholar] [CrossRef]
- Nonaka, G.; Ageta, M.; Nishioka, I. Tannins and related compounds. XXV. A new class of gallotannins possessing a (-)-shikimic acid core from Castanopsis cuspidata var. sieboldii NAKAI. (1). Chem. Pharm. Bull. 1985, 33, 96–101. [Google Scholar] [CrossRef]
- Ageta, M.; Nonaka, G.; Nishioka, I. Tannins and related compounds. LXVII.: Isolation and characterization of castanopsinins A-H, novel ellagitannins containing a triterpenoid glycoside core, from Castanopsis cuspidata var. sieboldii NAKAI. (3). Chem. Pharm. Bull. 1988, 36, 1646–1663. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Hatano, T. Constituents of Geranium thunbergii Sieb. et Zucc. Part 12. Hydrated stereostructure and equibration of geraniin. J. Chem. Soc. Perkin Trans. 1 1982, 9–14. [Google Scholar] [CrossRef]
- Tanaka, T.; Mine, C.; Watarumi, S.; Fujioka, T.; Mihashi, K.; Zhang, Y.; Kouno, I. Accumulation of epigallocatechin quinone dimers during tea fermentation and formation of theasinensins. J. Nat. Prod. 2002, 65, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nonaka, G.; Nishioka, I.; Ho, F. Bischofianin, a dimeric dehydroellagitannin from Bischofia javanica. Phytochemistry 1995, 38, 509–513. [Google Scholar] [CrossRef]
- Nandy, A.K.; Podder, G.; Sahu, N.P.; Mahato, S.B. Triterpenoids and their glucosides from Terminalia bellerica. Phytochemistry 1989, 28, 2769–2772. [Google Scholar] [CrossRef]
- Bisoli, E.; Garcez, W.S.; Hamerski, L.; Tieppo, C.; Garcez, F.R. Bioactive pentacyclic triterpenes from the stems of Combretum laxum. Molecules 2008, 13, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yoshida, T.; Hatano, T.; Koga, T.; Toh, N.; Kuriyama, K. Circular dichroism of hydrolysable tannins-I ellagitannins and gallotannins. Tetrahedron Lett. 1982, 23, 3937–3940. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yoshida, T.; Hatano, T.; Koga, T.; Toh, N.; Kuriyama, K. Circular dichroism of hydrolysable tannins-II dehydroellagitannins. Tetrahedron Lett. 1982, 23, 3941–3944. [Google Scholar] [CrossRef]
- Foo, L.Y. Amariin, a di-dehydrohexahydroxydiphenoyl hydrolysable tannin from Phyllanthus amarus. Phytochemistry 1993, 33, 487–491. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. J. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Sarotti, A.M. Computational chemistry to the rescue: Modern toolboxes for the assignment of complex molecules by GIAO NMR calculations. Chem. Eur. J. 2016, 22, 12246–12261. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Watarumi, S.; Matsuo, Y.; Kamei, M.; Kouno, I. Production of theasinensins A and D, epigallocatechin gallate dimers of black tea, by oxidation-reduction dismutation of dehydrotheasinensin A. Tetrahedron 2003, 59, 7939–7947. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 5.0.9; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1o | 1u | 2o a | |||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| Triterpene | ||||||
| 1 | 2.23 (m) | 47.0 | 2.27 (m) | 47.7 | 2.24 (m) | 47.3 |
| 1.53 (m) | 1.56 (m) | 1.54 (m) | ||||
| 2 | 4.46 (m) | 67.6 | 4.49 (m) | 67.8 | 4.48 (m) | 67.8 |
| 3 | 5.96 (d, 8.3) | 80.7 | 5.96 (d, 8.1) | 80.6 | 5.95 (d, 8.3) | 80.6 |
| 4 | 48.4 | 48.5 | 48.4 | |||
| 5 | 2.36 (m) | 45.7 | 2.37 (m) | 45.8 | 2.36 (m) | 45.8 |
| 6 | 1.90 (m) | 18.3 | 1.88 (m) | 18.5 | 1.92 (m) | 18.5 |
| 1.38 (m) | 1.43 (m) | |||||
| 7 | 1.72 (m) | 32.3 | 1.70 (m) | 33.3 | 1.67 (m) | 32.9 |
| 1.35 (m) | 1.33 (m) | |||||
| 8 | 39.8 | 40.2 | 40.0 | |||
| 9 | 1.90 (m) | 47.7 | 1.86 (m) | 47.8 | 1.90 (m) | 47.5 |
| 10 | 37.1 | 37.2 | 37.1 | |||
| 11 | 1.99 (m) | 24.0 | 2.01 (m) | 24.1 | 1.90 (2H, m) | 23.7 |
| 12 | 5.44 (m) | 122.6 | 5.46 (m) | 126.0 | 5.44 (m) | 122.4 |
| 13 | 143.8 | 138.5 | 144.8 | |||
| 14 | 42.1 | 42.7 | 42.3 | |||
| 15 | 1.08 (2H, m) | 28.0 | 1.02 (2H, m) | 30.0 | 1.12 (2H, m) | 28.2 |
| 16 | 1.91 (2H, m) | 22.7 | 1.99 (m) | 24.6 | 2.00 (2H, m) | 23.8 |
| 17 | 46.8 | 48.4 | 46.6 | |||
| 18 | 3.19 (m) | 41.6 | 2.50 (d, 11.3) | 53.4 | 3.26 (dd, 4.0, 13.7) | 42.0 |
| 19 | 1.21 (m) | 45.8 | 0.90 (m) | 39.1 | 1.76 (m) | 46.0 |
| 1.24 (m) | ||||||
| 20 | 30.5 | 39.3 | 32.7 | |||
| 21 | 1.04 (m) | 33.8 | 1.31 (m) | 30.8 | 1.18 (2H, m) | 34.1 |
| 22 | 1.23 (m) | 31.9 | 1.86 (m) | 36.8 | 1.93 (2H, m) | 37.3 |
| 1.71 (m) | ||||||
| 23 | 4.39 (m) | 62.4 | 4.42 (m) | 62.7 | 4.38 (d, 11.0) | 62.5 |
| 4.35 (m) | 4.17 (d, 11.0) | |||||
| 24 | 5.57 (d, 11.5) | 65.0 | 5.57 (d, 11.7) | 65.2 | 5.56 (d, 11.7) | 65.1 |
| 4.06 (m) | 4.04 (d, 11.5) | 4.06 (d, 11.7) | ||||
| 25 | 1.00 (3H, s) | 23.2 | 1.04 (3H, s) | 17.0 | 0.98 (3H, s) | 17.4 |
| 26 | 1.10 (3H, s) | 17.3 | 1.14 (3H, s) | 17.8 | 1.01 (3H, s) | 16.9 |
| 27 | 1.14 (3H, s) | 25.8 | 1.09 (3H, s) | 23.7 | 1.12 (3H, s) | 26.0 |
| 28 | 176.2 | 176.2 | 180.1 | |||
| 29 | 0.87 (3H, s) | 32.9 | 0.90 (3H, d, 6.3) | 17.3 | 0.91 (3H, s) | 33.2 |
| 30 | 0.86 (3H, s) | 23.4 | 0.86 (3H, s) | 21.2 | 0.97 (3H, s) | 23.6 |
| Glucose | ||||||
| 1 | 6.31 (d, 8.0) | 95.6 | 6.25 (d, 8.1) | 95.7 | ||
| 2 | 4.19 (m) | 74.0 | 4.19 (m) | 74.1 | ||
| 3 | 4.27 (m) | 78.7 | 4.27 (m) | 78.9 | ||
| 4 | 4.37 (m) | 70.9 | 4.38 (m) | 71.2 | ||
| 5 | 4.02 (m) | 79.1 | 4.01 (m) | 79.2 | ||
| 6 | 4.45 (m) | 62.0 | 4.20 (m) | 62.3 | ||
| Phenazine | ||||||
| 1 | 117.2 | 117.5 | 117.4 | |||
| 2 | 136.6 | 135.1 | 136.0 | |||
| 3 | 8.30(s) | 118.2 | 8.29 (s) | 118.3 | 8.13 (s) | 118.3 |
| 4 | 142.3 | 142.4 | 142.4 | |||
| 5 | 136.3 | 136.5 | 136.5 | |||
| 6 | 153.3 | 153.5 | 153.4 | |||
| 7 | 169.1 | 169.2 | 169.2 | |||
| 9 | 142.1 b | 142.1 e | 143.9 h | |||
| 10 | 7.70 (m) | 130.4 c | 7.71 (s) | 130.6 f | 7.72 (m) | 130.6 i |
| 11 | 8.12 (m) | 129.7 d | 8.13 (m) | 129.5 g | 8.06 (m) | 129.8 j |
| 12 | 8.17 (m) | 129.3 d | 8.18 (m) | 129.9 g | 8.12 (m) | 129.4 j |
| 13 | 7.70 (m) | 130.3 c | 7.71 (m) | 130.5 f | 7.71 (m) | 130.4 i |
| 14 | 143.9 b | 144.0 e | 142.0 h | |||
| 1’ | 115.5 | 115.6 | 115.6 | |||
| 2’ | 125.0 | 125.2 | 125.5 | |||
| 3’ | 7.49 (s) | 108.5 | 7.49 (s) | 108.7 | 7.50 (s) | 108.6 |
| 4’ | 147.1 | 147.3 | 147.2 | |||
| 5’ | 138.6 | 138.8 | 138.7 | |||
| 6’ | 146.8 | 147.0 | 146.7 | |||
| 7’ | 169.2 | 169.4 | 169.3 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakamatsu, H.; Tanaka, S.; Matsuo, Y.; Saito, Y.; Nishida, K.; Tanaka, T. Reductive Metabolism of Ellagitannins in the Young Leaves of Castanopsis sieboldii. Molecules 2019, 24, 4279. https://doi.org/10.3390/molecules24234279
Wakamatsu H, Tanaka S, Matsuo Y, Saito Y, Nishida K, Tanaka T. Reductive Metabolism of Ellagitannins in the Young Leaves of Castanopsis sieboldii. Molecules. 2019; 24(23):4279. https://doi.org/10.3390/molecules24234279
Chicago/Turabian StyleWakamatsu, Hatsumi, Sumire Tanaka, Yosuke Matsuo, Yoshinori Saito, Koyo Nishida, and Takashi Tanaka. 2019. "Reductive Metabolism of Ellagitannins in the Young Leaves of Castanopsis sieboldii" Molecules 24, no. 23: 4279. https://doi.org/10.3390/molecules24234279
APA StyleWakamatsu, H., Tanaka, S., Matsuo, Y., Saito, Y., Nishida, K., & Tanaka, T. (2019). Reductive Metabolism of Ellagitannins in the Young Leaves of Castanopsis sieboldii. Molecules, 24(23), 4279. https://doi.org/10.3390/molecules24234279