
Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
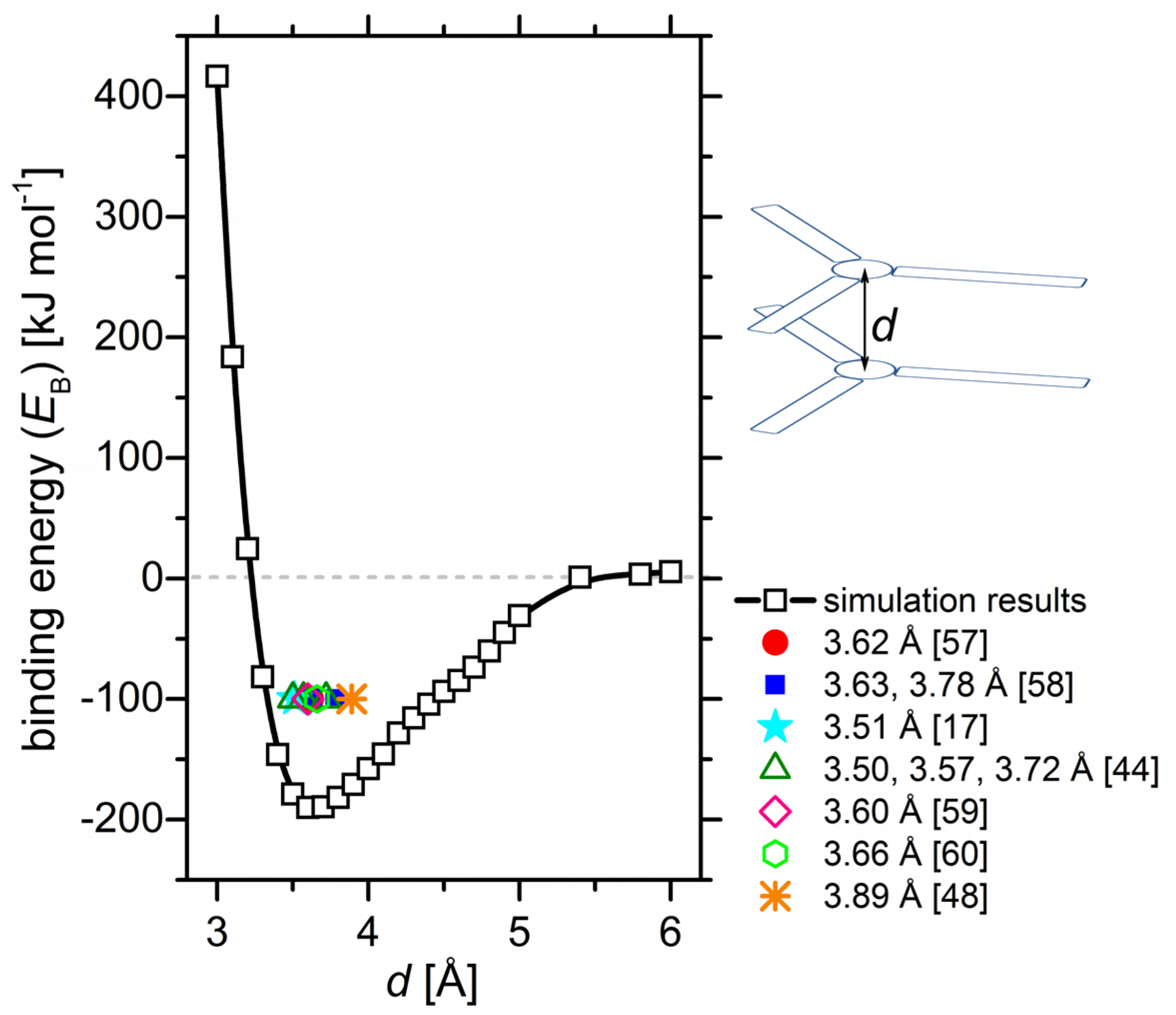
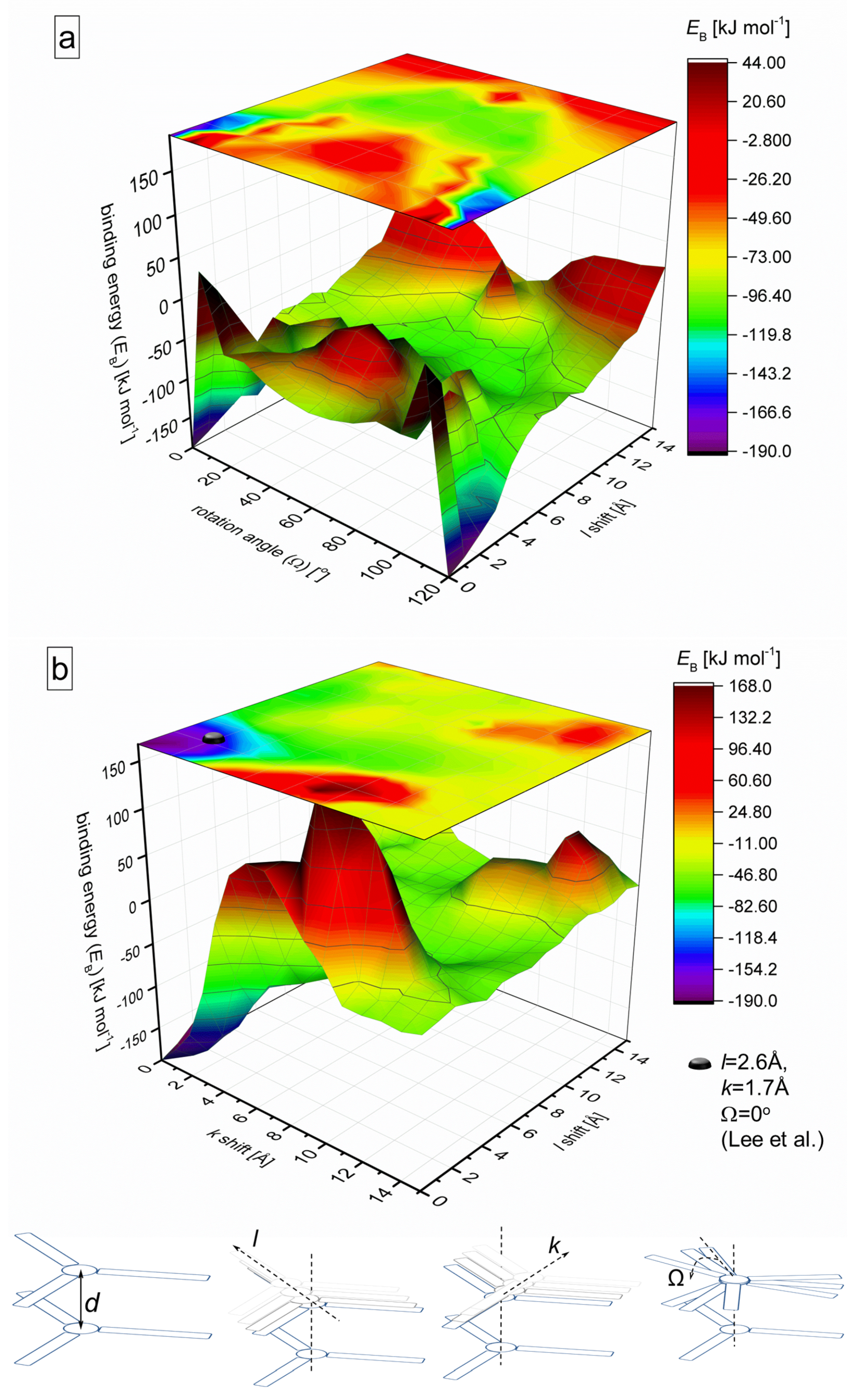
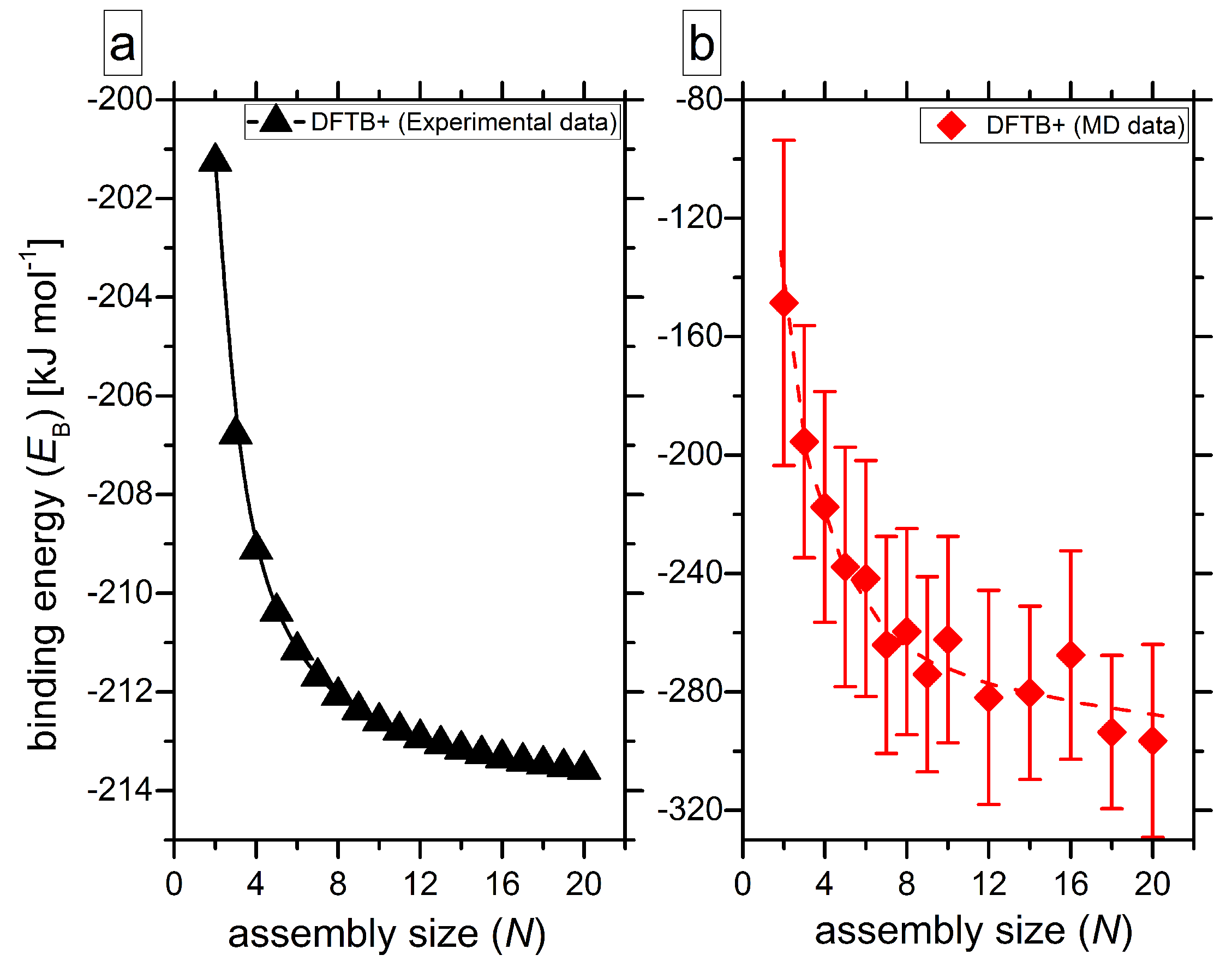
2.1. Binding Energy Diagrams
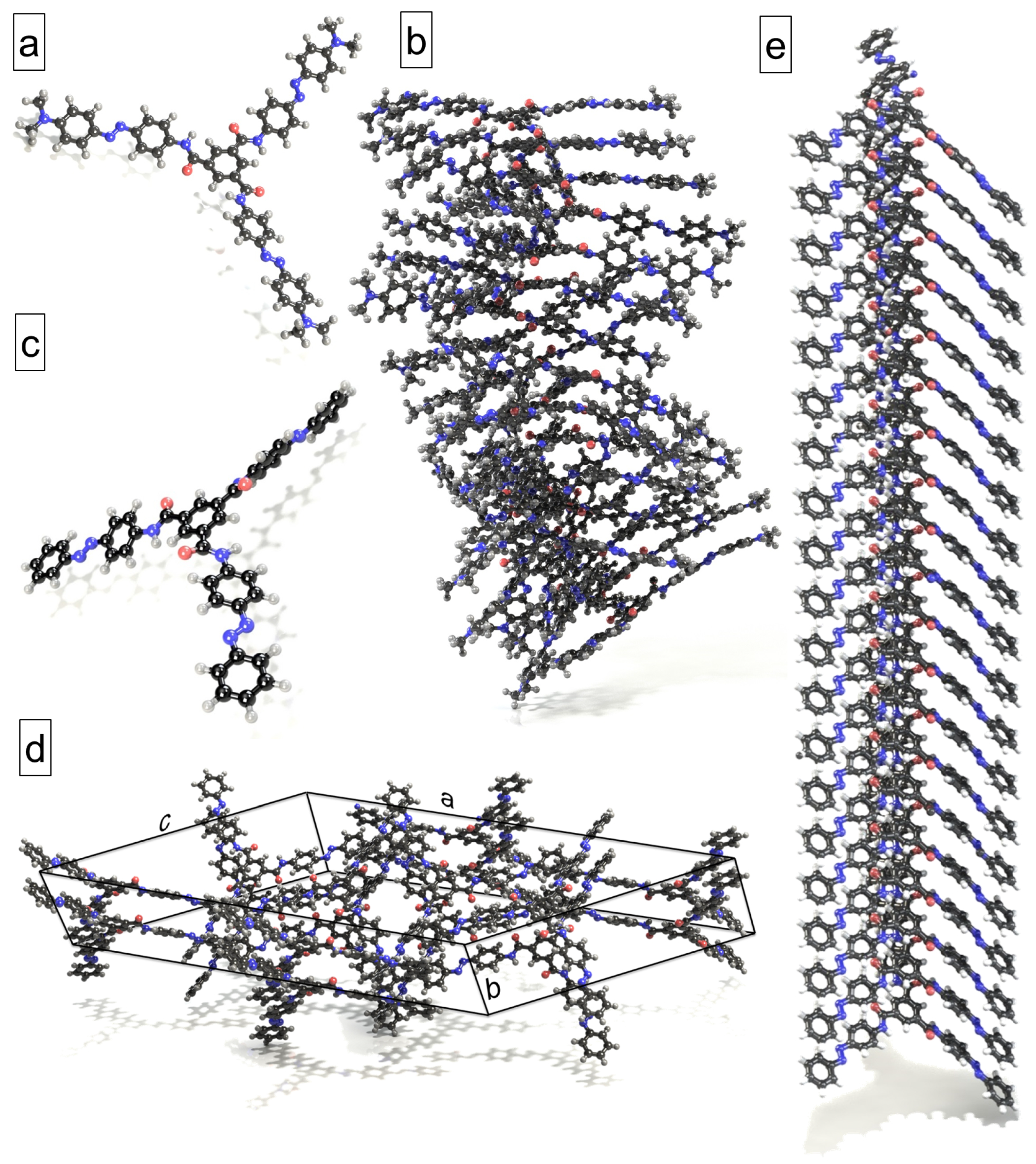
2.2. Analysis of the Trans-Azo–BTA Star Crystal
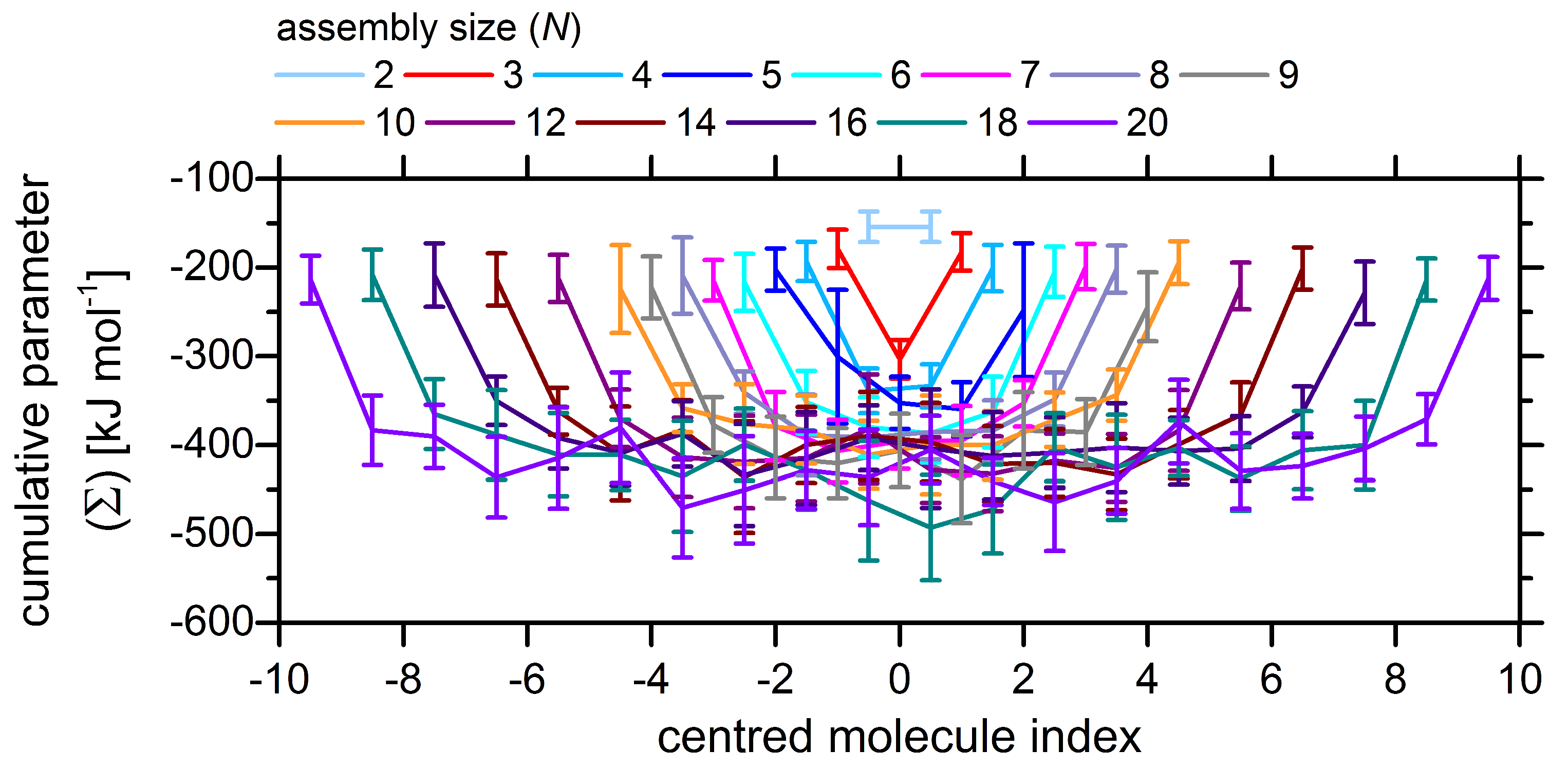
2.3. On the Cooperative Nature of Self-Assembly
3. Conclusions
4. Materials and Methods
4.1. Objects of the Study and Their Models
4.2. Computation of the Binding Energy Diagrams and the Binding Energy as a Function of Assembly Size N
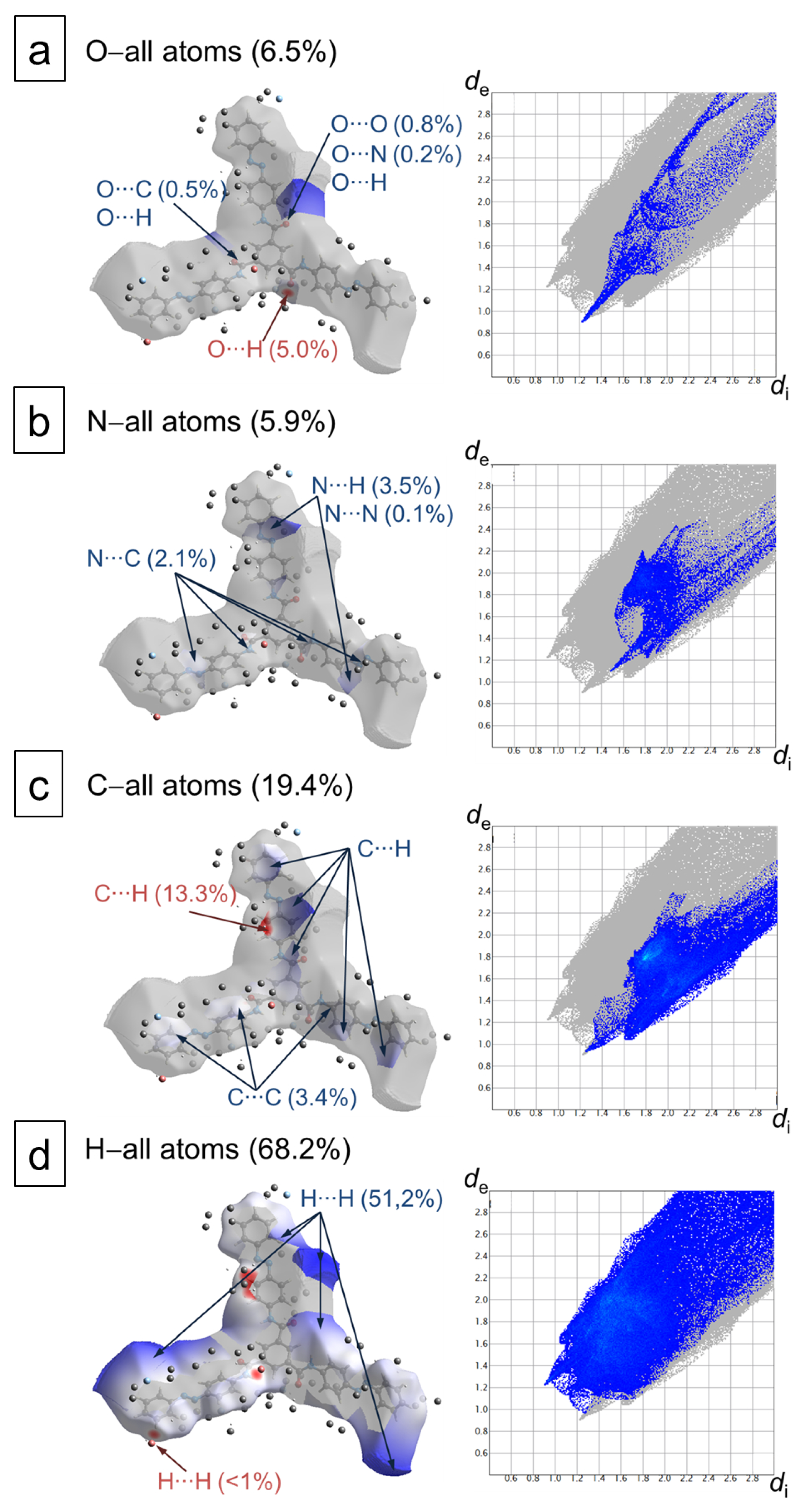
4.3. Hirshfeld Surface Analysis of the azo–BTA Star Crystal
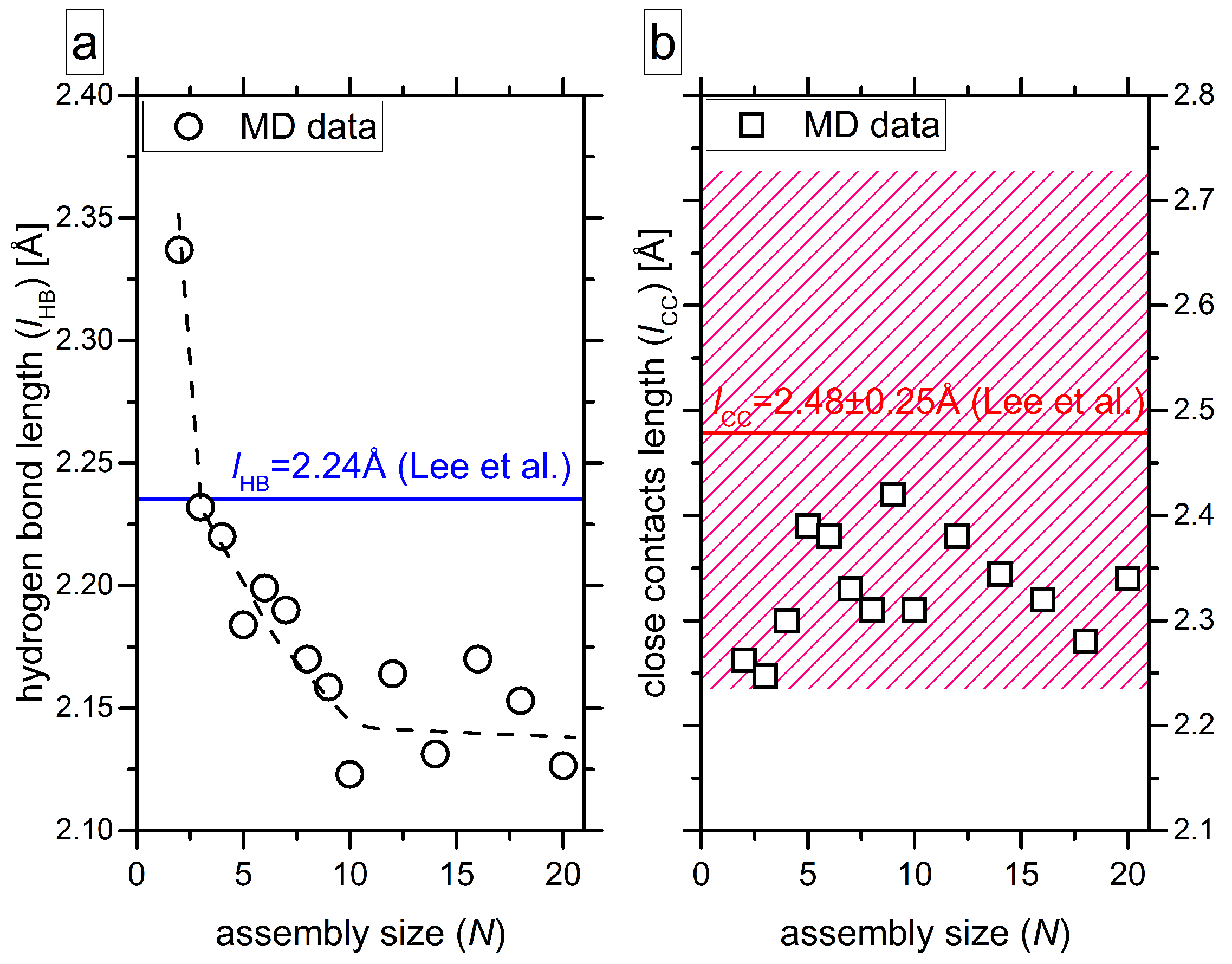
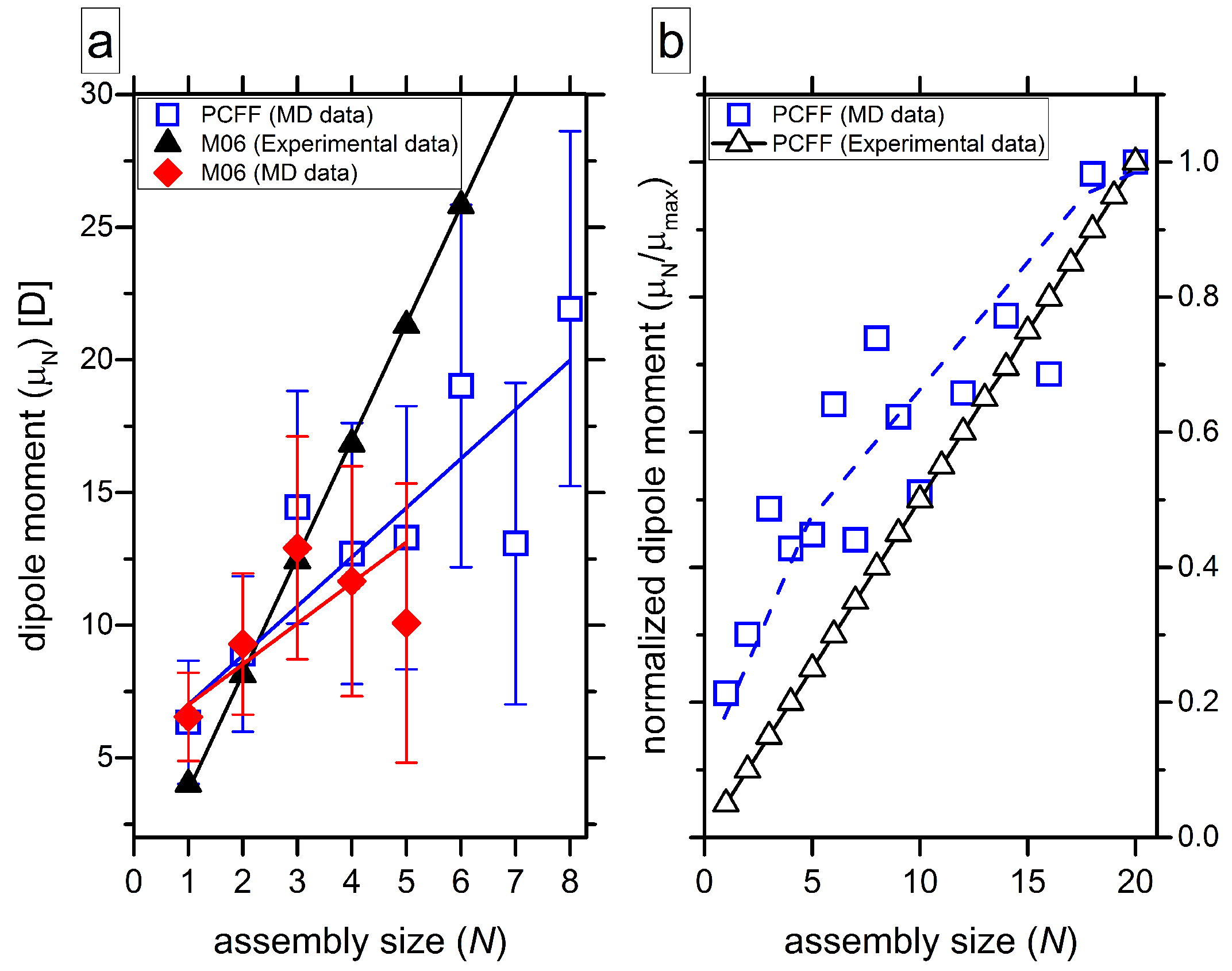
4.4. Quantification of Hydrogen Bonds, Close Contacts and Macrodipoles
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BTA | Benzene-1,3,5-tricarboxamide |
| azo–BTA star | Three-arm star-shaped molecule with BTA core and azobenzene arms |
| DFT | Density functional theory |
| DFTB+ | Density functional based tight-binding |
| MD | Molecular dynamics |
| PCFF | Polymer consistent force field |
| LAMMPS | Large-scale atomic/molecular massively parallel simulator |
References
- Ikeda, T.; Tsutsumi, O. Optical switching and image storage by means of azobenzene liquid-crystal films. Science 1995, 268, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.A.; Wooley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef]
- Gillono, M.; Roppolo, I.; Frascella, F.; Scaltrito, L.; Pirri, C.F.; Chiappone, A. CO2 permeability control in 3D printed light responsive structures. Appl. Mater. Today 2019, 100470. [Google Scholar] [CrossRef]
- Serak, S.V.; Tabiryan, N.V.; White, T.J.; Bunning, T.J. Azobenzene liquid crystal polymer-based membrane and cantilever optical systems. Opt. Express 2009, 17, 15736–15746. [Google Scholar] [CrossRef] [PubMed]
- Jelken, J.; Pandiyarajan, C.K.; Genzer, J.; Lomadze, N.; Santer, S. Fabrication of flexible hydrogel sheets featuring periodically spaced holes with continuously adjustable size in real time. ACS Appl. Mater. Interfaces 2018, 10, 30844–30851. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, Y.; Lv, J.-M.; Min, Y.; Quan, Y.-Y.; Ge, L.-N.; Tian, M.; Yao, D.-S. Multi-arm azobenzene liquid crystal based on cholic acid: Synthesis and mesophase properties. Liq. Cryst. 2018, 45, 1813–1824. [Google Scholar] [CrossRef]
- Vialetto, J.; Groppi, J.; La Rosa, M.; Silvi, S.; Credi, A.; Baroncini, M. Solution and solid state photochromism in a family of shape persistent azobenzene tetramers functionalized with alkyloxy substituents. Photochem. Photobiol. Sci. 2019, 18, 2281–2286. [Google Scholar] [CrossRef]
- Ooi, Y.-H.; Yeap, G.-Y.; Han, C.-C.; Lin, H.-C.; Shinomiya, T.; Ito, M.M. Non-conventional three-armed star-shaped mesogens based on 1, 3, 5-trisubstituted benzene with azobenzene moieties at the periphery: Synthesis, and mesomorphic behaviour. Liq. Cryst. 2014, 41, 1017–1033. [Google Scholar] [CrossRef]
- Takahashi, T.; Tanino, T.; Ando, H.; Nakano, H.; Shirota, Y. Surface relief grating formation using a novel azobenzene-based photochromic amorphous molecular material, tris [4-(phenylazo) phenyl] amine. Mol. Cryst. Liq. Cryst. 2005, 430, 9–14. [Google Scholar] [CrossRef]
- Yadavalli, N.S.; Saphiannikova, M.; Santer, S. Photosensitive response of azobenzene containing films towards pure intensity or polarization interference patterns. Appl. Phys. Lett. 2014, 105, 051601. [Google Scholar] [CrossRef]
- Jiang, X.; Lu, J.; Zhou, F.; Zhang, Z.; Pan, X.; Zhang, W.; Wang, Y.; Zhou, N.; Zhu, X. Molecularly-defined macrocycles containing azobenzene main-chain oligomers: Modular stepwise synthesis, chain-length and topology-dependent properties. Polym. Chem. 2016, 7, 2645–2651. [Google Scholar] [CrossRef]
- Imrie, C.T.; Henderson, P.A. Liquid crystal dimers and higher oligomers: Between monomers and polymers. Chem. Soc. Rev. 2007, 36, 2096–2124. [Google Scholar] [CrossRef] [PubMed]
- Imrie, C.T.; Henderson, P.; Yeap, G.-Y. Liquid crystal oligomers: Going beyond dimers. Liq. Cryst. 2009, 36, 755–777. [Google Scholar] [CrossRef]
- Koch, M.; Saphiannikova, M.; Santer, S.; Guskova, O. Photoisomers of azobenzene star with a flat core: Theoretical insights into multiple states from DFT and MD perspective. J. Phys. Chem. B 2017, 121, 8854–8867. [Google Scholar] [CrossRef]
- Galanti, A.; Santoro, J.; Mannancherry, R.; Duez, Q.; Diez-Cabanes, V.; Valášek, M.; De Winter, J.; Cornil, J.; Gerbaux, P.; Mayor, M.; et al. A new class of rigid multi (azobenzene) switches featuring electronic decoupling: Unravelling the isomerization in individual photochromes. J. Am. Chem. Soc. 2019, 141, 9273–9283. [Google Scholar] [CrossRef]
- Han, M.; Nakanishi, H. Light-sensitive microspheres based on spherical assembly of star-shaped chromophores. J. Photopolym. Sci. Technol. 2018, 31, 527–531. [Google Scholar] [CrossRef]
- Lee, S.; Oh, S.; Lee, J.; Malpani, Y.; Jung, Y.-S.; Kang, B.; Lee, J.Y.; Ozasa, K.; Isoshima, T.; Lee, S.Y.; et al. Stimulus-responsive azobenzene supramolecules: Fibers, gels, and hollow spheres. Langmuir 2013, 29, 5869–5877. [Google Scholar] [CrossRef]
- Lee, J.; Oh, S.; Pyo, J.; Kim, J.-M.; Je, J.H. A light-driven supramolecular nanowire actuator. Nanoscale 2015, 7, 6457–6461. [Google Scholar] [CrossRef]
- Bahrenburg, J.; Sievers, C.M.; Schönborn, J.B.; Hartke, B.; Renth, F.; Temps, F.; Näther, C.; Sönnichsen, F.D. Photochemical properties of multi-azobenzene compounds. Photochem. Photobiol. Sci. 2013, 12, 511–518. [Google Scholar] [CrossRef]
- Adachi, H.; Hirai, Y.; Ikeda, T.; Maeda, M.; Hori, R.; Kutsumizu, S.; Haino, T. Photoresponsive toroidal nanostructure formed by self-assembly of azobenzene-functionalized tris(phenylisoxazolyl) benzene. Org. Lett. 2016, 18, 924–927. [Google Scholar] [CrossRef]
- Baroncini, M.; d’Agostino, S.; Bergamini, G.; Ceroni, P.; Comotti, A.; Sozzani, P.; Bassanetti, I.; Grepioni, F.; Hernande, T.M.; Silvi, S.; et al. Photoinduced reversible switching of porosity in molecular crystals based on star-shaped azobenzene tetramers. Nat. Chem. 2015, 7, 634. [Google Scholar] [CrossRef] [PubMed]
- Nacci, C.; Baroncini, M.; Credi, A.; Grill, L. Reversible photoswitching and isomer-dependent diffusion of single azobenzene tetramers on a metal surface. Angew. Chem. Int. Ed. 2018, 130, 15254–15259. [Google Scholar] [CrossRef]
- Akiyama, H. Photochemically reversible liquefaction/solidification of sugar–alcohol derivatives. In Functional Organic Liquids; Wiley–VCH Verlag GmbH Co.: Weinheim, Germany, 2019; pp. 75–86. [Google Scholar]
- Liao, L.-X.; Junge, D.M.; McGrath, D.V. Photochromic dendrimers containing six azobenzenes. Macromolecules 2002, 35, 319–322. [Google Scholar] [CrossRef]
- Tanaka, D.; Ishiguro, H.; Shimizu, Y.; Uchida, K. Thermal and photoinduced liquid crystalline phase transitions with a rod–disc alternative change in the molecular shape. J. Mater. Chem. 2012, 22, 25065–25071. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Kim, D.-Y.; Park, M.; Yoon, W.-J.; Lee, Y.; Hwang, J.-K.; Chiang, Y.-W.; Kuo, S.-W.; Hsu, C.-H.; Jeong, K.-U. Self-assembled hierarchical superstructures from the benzene-1, 3, 5-tricarboxamide supramolecules for the fabrication of remote-controllable actuating and rewritable films. ACS Appl. Mater. Interfaces 2016, 8, 9490–9498. [Google Scholar] [CrossRef] [PubMed]
- Segarra-Maset, M.D.; van Leeuwen, P.W.N.M.; Freixa, Z. Light switches the ligand! Photochromic azobenzene–phosphanes. Eur. J. Inorg. Chem. 2010, 2010, 2075–2078. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Q.; Zhang, J.; Hou, W. Photochemical behaviours of star-like liquid crystal with azobenzene terminal groups. J. Mater. Sci. 2005, 40, 4517–4521. [Google Scholar] [CrossRef]
- Koch, M.; Saphiannikova, M.; Guskova, O. Do columns of azobenzene stars disassemble under light illumination? Langmuir 2019, 35, 14659–14669. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, P.; Audorff, H.; Kreger, K.; Kador, L.; Schmidt, H.-W.; Stingelin, N.; Smith, P. Photo-induced molecular alignment of trisazobenzene derivatives. J. Mater. Chem. 2011, 21, 4339–4345. [Google Scholar] [CrossRef]
- Wolfer, P.; Kreger, K.; Schmidt, H.-W.; Stingelin, N.; Smith, P. Photo-oriented trisazobenzene layers for patterned liquid-crystal alignment. Mol. Cryst. Liq. Cryst. 2012, 562, 133–140. [Google Scholar] [CrossRef]
- Kreger, K.; Wolfer, P.; Audorff, H.; Kador, L.; Stingelin-Stutzmann, N.; Smith, P.; Schmidt, H.-W. Stable holographic gratings with small-molecular trisazobenzene derivatives. J. Am. Chem. Soc. 2010, 132, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Zhou, Y.; Wang, X.; He, Y. Fast photoinduced large deformation of colloidal spheres from a novel 4-arm azobenzene compound. ACS Appl. Mater. Interfaces 2015, 7, 16889–16895. [Google Scholar] [CrossRef] [PubMed]
- Sperner, M.; Tober, N.; Detert, H. Tristriazolotriazines with azobenzene arms-acidochromic dyes and discotic liquid crystals. Eur. J. Org. Chem. 2019, 2019, 4688–4693. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, D.; Janietz, D.; Schmidt, C.; Wendorff, J.H. Liquid crystalline 1, 3, 5-triazines incorporating rod-like azobenzene sub-units. Liq. Cryst. 1998, 25, 711–719. [Google Scholar] [CrossRef]
- Pokladek, Z.; Dudek, M.; Mongin, O.; Metivier, R.; Mlynarz, P.; Samoc, M.; Matczyszyn, K.; Paul, F. Linear and third-order nonlinear optical properties of triazobenzene-1, 3, 5-triazinane-2, 4, 6-trione (isocyanurate) derivatives. ChemPlusChem 2017, 82, 1372–1383. [Google Scholar] [CrossRef]
- Wu, S.L.; Zhao, F.Y.; Luo, S.W.; Zhang, W.J.; Xu, S.D.; Zeng, Q.D. Synthesis and scanning tunneling microscopy observation of 1, 3, 5-triazine bearing azo-carboxylate. J. Nanosci. Nanotechnol. 2017, 17, 725–728. [Google Scholar] [CrossRef]
- Jin, H.; Jian, T.; Ding, Y.-H.; Chen, Y.; Mu, P.; Wang, L.; Chen, C.-L. Solid-phase synthesis of three-armed star-shaped peptoids and their hierarchical self-assembly. Biopolymers 2019, 110, e23258. [Google Scholar] [CrossRef]
- Vera, F.; Tejedor, R.M.; Romero, P.; Barberá, J.; Ros, M.B.; Serrano, J.L.; Sierra, T. Light-driven supramolecular chirality in propeller-like hydrogen-bonded complexes that show columnar mesomorphism. Angew. Chem. Int. Ed. 2007, 46, 1873–1877. [Google Scholar] [CrossRef]
- Metzroth, T.; Hoffmann, A.; Martín-Rapún, R.; Smulders, M.M.J.; Pieterse, K.; Palmans, A.R.A.; Vekemans, J.A.J.M.; Meijer, E.W.; Spiess, H.W.; Gauss, J. Unravelling the fine structure of stacked bipyridine diamine-derived C3-discotics as determined by X–ray diffraction, quantum-chemical calculations, fast-MAS NMR and CD spectroscopy. Chem. Sci. 2011, 2, 69–76. [Google Scholar] [CrossRef]
- Van Herrikhuyzen, J.; Jonkheijm, P.; Schenning, A.P.H.J.; Meijer, E.W. The influence of hydrogen bonding and π–π stacking interactions on the self-assembly properties of C3–symmetrical oligo (p-phenylenevinylene) discs. Org. Biomol. Chem. 2006, 4, 1539–1545. [Google Scholar] [CrossRef]
- Narayan, B.; Kulkarni, C.; George, S.J. Synthesis and self-assembly of a C3–symmetric benzene-1, 3, 5-tricarboxamide (BTA) anchored naphthalene diimide disc. J. Mater. Chem. C 2013, 1, 626–629. [Google Scholar] [CrossRef]
- Smulders, M.M.J.; Schenning, A.P.H.J.; Meijer, E.W. Insight into the mechanisms of cooperative self-assembly: The “sergeants-and-soldiers” principle of chiral and achiral C3–symmetrical discotic triamides. J. Am. Chem. Soc. 2008, 130, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Filot, I.A.W.; Palmans, A.R.A.; Hilbers, P.A.J.; van Santen, R.A.; Pidko, E.A.; de Greef, T.F.A. Understanding cooperativity in hydrogen-bond-induced supramolecular polymerization: A density functional theory study. J. Phys. Chem. B 2010, 114, 13667–13674. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, C.; Meijer, E.W.; Palmans, A.R.A. Cooperativity scale: A structure–mechanism correlation in the self-assembly of benzene-1, 3, 5-tricarboxamides. Acc. Chem. Res. 2017, 50, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, D.; Pavan, G.M. Molecular modelling of supramolecular polymers. Adv. Phys. X 2018, 3, 316–338. [Google Scholar] [CrossRef] [Green Version]
- Malpani, Y.R.; Oh, S.; Lee, S.; Jung, Y.-S.; Kim, J.-M. Photoinduced phase transition of azobenzene-coupled benzenetricarboxamide. Bull. Korean Chem. Soc 2014, 35, 2563–2566. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Bala, I.; Gupta, S.P.; Kumar, P.; Pal, S.K.; Venkataramani, S. Reversibly photoswitchable alkoxy azobenzenes connected benzenetricarboxamide discotic liquid crystals with perpetual long range columnar assembly. Org. Biomol. Chem. 2019, 17, 1947–1954. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Yoon, W.-J.; Park, M.; Kang, D.-G.; Bang, G.; Koo, J.; Lim, S.-I.; Park, S.; Jeong, K.-U. Construction of light-responsive phase chirality from an achiral macrogelator. J. Mater. Chem. C 2019, 7, 3231–3237. [Google Scholar] [CrossRef]
- Leyendecker, M. Supramolecular Lyotropic Liquid Crystalline Alignment Media Based on Benzene-1,3,5-tricarboxamides; Technische Universität Darmstadt: Darmstadt, Germany, 2019. [Google Scholar]
- Huang, Y.; Kim, D.-H. Light-controlled synthesis of gold nanoparticles using a rigid, photoresponsive surfactant. Nanoscale 2012, 4, 6312–6317. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Kulikovsky, L.; Kulikovska, O.; Tomczyk, J.; Stumpe, J. Thin layers of low molecular azobenzene materials with effective light-induced mass transport. Langmuir 2010, 26, 2214–2217. [Google Scholar] [CrossRef]
- Yadavalli, N.S. Advances in Experimental Methods to Probe Surface Relief Grating Formation Mechanism in Photosensitive Materials; Universität Potsdam: Potsdam, Germany, 2014. [Google Scholar]
- Wolfer, P. Ordering Molecular Organics into Functionality; ETH Zürich: Zürich, Germany, 2011. [Google Scholar]
- Taniguchi, T.; Asahi, T.; Koshima, H. Photomechanical azobenzene crystals. Crystals 2019, 9, 437. [Google Scholar] [CrossRef] [Green Version]
- De Greef, T.F.A.; Smulders, M.M.J.; Wolffs, M.; Schenning, A.P.H.J.; Sijbesma, R.P.; Meijer, E.W. Supramolecular polymerization. Chem. Rev. 2009, 109, 5687–5754. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, M.P.; Mair, F.S.; Pritchard, R.G.; Warren, J.E. New supramolecular packing motifs: π-stacked rods encased in triply-helical hydrogen bonded amide strands. Chem. Commun. 1999, 1945–1946. [Google Scholar] [CrossRef]
- Nakano, Y.; Hirose, T.; Stals, P.J.M.; Meijer, E.W.; Palmans, A.R.A. Conformational analysis of supramolecular polymerization processes of disc-like molecules. Chem. Sci. 2012, 3, 148–155. [Google Scholar] [CrossRef]
- Ranganathan, D.; Kurur, S.; Gilardi, R.; Karle, I.L. Design and synthesis of AB3-type (A = 1,3,5-benzenetricarbonyl unit; B = Glu diOMe or Glu7 octa OMe) peptide dendrimers: Crystal structure of the first generation. Biopolym. Orig. Res. Biom. 2000, 54, 289–295. [Google Scholar] [CrossRef]
- Gong, B.; Zheng, C.; Yan, Y. Structure of N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide trihydrate. J. Chem. Crystallogr. 1999, 29, 649–652. [Google Scholar] [CrossRef]
- Albuquerque, R.Q.; Timme, A.; Kress, R.; Senker, J.; Schmidt, H.-W. Theoretical investigation of macrodipoles in supramolecular columnar stackings. Chem. Eur. J. 2013, 19, 1647–1657. [Google Scholar] [CrossRef]
- Wegner, M.; Dudenko, D.; Sebastiani, D.; Palmans, A.R.A.; de Greef, T.F.A.; Graf, R.; Spiess, H.W. The impact of the amide connectivity on the assembly and dynamics of benzene-1,3,5-tricarboxamides in the solid state. Chem. Sci. 2011, 2, 2040–2049. [Google Scholar] [CrossRef]
- Greciano, E.E.; Calbo, J.; Buendía, J.; Cerdá, J.; Aragó, J.; Ortí, E.; Sánchez, L. Decoding the consequences of increasing the size of self-assembling tricarboxamides on chiral amplification. J. Am. Chem. Soc. 2019, 141, 7463–7472. [Google Scholar] [CrossRef]
- Korlepara, D.B.; Henderson, W.R.; Castellano, R.K.; Balasubramanian, S. Differentiating the mechanism of self-assembly in supramolecular polymers through computation. Chem. Commun. 2019, 55, 3773–3776. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Garzoni, M.; Baker, M.B.; Leenders, C.M.A.; Voets, I.K.; Albertazzi, L.; Palmans, A.R.A.; Meijer, E.W.; Pavan, G.M. Effect of H–bonding on order amplification in the growth of a supramolecular polymer in water. J. Am. Chem. Soc. 2016, 138, 13985–13995. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, D.; Kwangmettatam, S.; Kudernac, T.; Pavan, G.M. How defects control the out-of-equilibrium dissipative evolution of a supramolecular tubule. ACS Nano 2019, 13, 4322–4334. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennicci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA Materials Studio. Dassault Systéms; Version 8; BIOVIA Materials Studio: San Diego, CA, USA, 2014. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B Condens. Matter Phys. 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer (Version 3.1); University of Western Australia: Perth, WA, Australia, 2012. [Google Scholar]
- Cruickshank, D.L.; Hendon, C.H.; Verbeek, M.J.R.; Walsh, A.; Wilson, C.C. Polymorphism of the azobenzene dye compound methyl yellow. CrystEngComm 2016, 18, 3456–3461. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, M.; Uchida, E.; Norikane, Y.; Azumi, R.; Nozawa, S.; Tomita, A.; Sato, T.; Adachi, S.-I.; Koshihara, S.-Y. Crystal melting by light: X-ray crystal structure analysis of an azo crystal showing photoinduced crystal–melt transition. J. Am. Chem. Soc. 2014, 136, 9158–9164. [Google Scholar] [CrossRef]
- Scott, J.L.; Almesaker, A.; Sumi, Y.; Tanaka, K. Guest signaling compound: Trans-3,3′-bis(diphenylhydroxymethyl)azobenzene. Cryst. Growth Des. 2007, 7, 1049–1054. [Google Scholar] [CrossRef]
- Scott, J.L.; Tanaka, K. Signalling by modulation of intermolecular interactions. In Engineering of Crystalline Materials Properties. NATO Science for Peace and Security Series B: Physics and Biophysics; Springer: Dordrecht, The Netherlands, 2008; pp. 429–447. [Google Scholar]
- Luzar, A.; Chandler, D. Hydrogen-bond kinetics in liquid water. Nature 1996, 379, 55–57. [Google Scholar] [CrossRef]
- Zawada, A.; Kaczmarek-Kędziera, A.; Bartkowiak, W. On the potential application of DFT methods in predicting the interaction-induced electric properties of molecular complexes. Molecular H-bonded chains as a case of study. J. Mol. Model. 2012, 18, 3073–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni–j | N1–2 | N2–3 | N3–4 | N4–5 | N5–6 | N6–7 | N7–8 | N8–9 | N9–10 | |
| lHB | 2.34 | 2.14 | 2.16 | 2.22 | 2.15 | 1.93 | 2.15 | 2.00 | 2.02 | |
| Ni–j | N10–11 | N11–12 | N12–13 | N13–14 | N14–15 | N15–16 | N16–17 | N17–18 | N18–19 | N19–20 |
| lHB | 2.18 | 2.08 | 2.17 | 2.05 | 1.99 | 2.04 | 2.11 | 2.08 | 2.21 | 2.25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savchenko, V.; Koch, M.; Pavlov, A.S.; Saphiannikova, M.; Guskova, O. Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling. Molecules 2019, 24, 4387. https://doi.org/10.3390/molecules24234387
Savchenko V, Koch M, Pavlov AS, Saphiannikova M, Guskova O. Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling. Molecules. 2019; 24(23):4387. https://doi.org/10.3390/molecules24234387
Chicago/Turabian StyleSavchenko, Vladyslav, Markus Koch, Aleksander S. Pavlov, Marina Saphiannikova, and Olga Guskova. 2019. "Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling" Molecules 24, no. 23: 4387. https://doi.org/10.3390/molecules24234387
APA StyleSavchenko, V., Koch, M., Pavlov, A. S., Saphiannikova, M., & Guskova, O. (2019). Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling. Molecules, 24(23), 4387. https://doi.org/10.3390/molecules24234387