pKa Determination of a Histidine Residue in a Short Peptide Using Raman Spectroscopy


Abstract
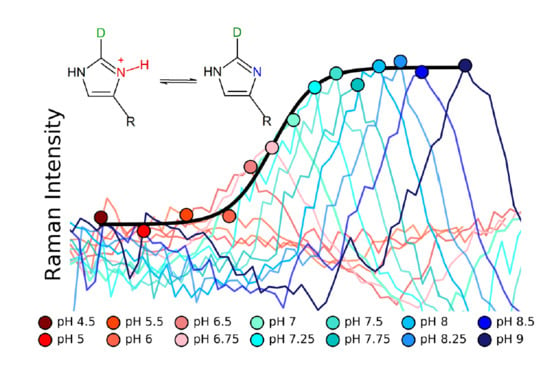
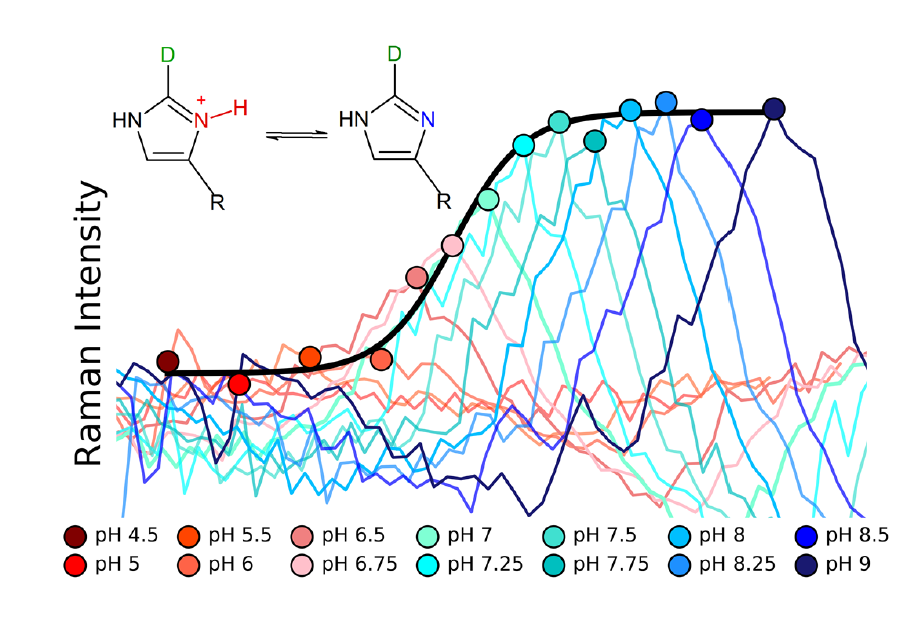
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. HVD Deuterium Exchange Reaction
2.2. pKa Determination
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Peptide Synthesis and Purification
4.3. Deuterium Exchange Reaction
4.4. Raman Sample Preparation
4.5. Raman Spectroscopy
4.6. Raman Data Processing
4.7. 1H NMR Spectroscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A1. Materials
Appendix A2. Peptide Synthesis and Purification
References
- Adhikary, R.; Zimmermann, J.; Romesberg, F.E. Transparent Window Vibrational Probes for the Characterization of Proteins with High Structural and Temporal Resolution. Chem. Rev. 2017, 117, 1927–1969. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cho, M. Infrared Probes for Studying the Structure and Dynamics of Biomolecules. Chem. Rev. 2013, 113, 5817–5847. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pazos, I.M.; Zhang, W.; Culik, R.M.; Gai, F. Site-Specific Infrared Probes of Proteins. Annu. Rev. Phys. Chem. 2015, 66, 357–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostron, P.; Garber, D.; Gaber, S. Raman Spectroscopy, a review. Int. J. Eng. Tech. Res. 2016, 6, 50–64. [Google Scholar]
- Chin, J.K.; Jimenez, R.; Romesberg, F.E. Direct Observation of Protein Vibrations by Selective Incorporation of Spectroscopically Observable Carbon−Deuterium Bonds in Cytochrome c. J. Am. Chem. Soc. 2001, 123, 2426–2427. [Google Scholar] [CrossRef] [PubMed]
- Thielges, M.C.; Case, D.A.; Romesberg, F.E. Carbon−Deuterium Bonds as Probes of Dihydrofolate Reductase. J. Am. Chem. Soc. 2008, 130, 6597–6603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noether, H.D. Infra-Red and Raman Spectra of Polyatomic Molecules XVII. Methyl-d3-Alcohol-d and Methyl-d3-Alcohol. J. Chem. Phys. 1942, 10, 693–699. [Google Scholar] [CrossRef]
- Neelakantan, P.; Krishnan, R.S.; Iitaka, Y. Raman spectrum of C-deuterated γ-glycine (NH3+CD2COO−). Proc. Indian Acad. Sci. Sect. A 1963, 58, 275–278. [Google Scholar] [CrossRef]
- Sunder, S.; Mendelsohn, R.; Bernstein, H.J. Raman studies of the C-H and C-D stretching regions in stearic acid and some specifically deuterated derivatives. Chem. Phys. Lipids 1976, 17, 456–465. [Google Scholar] [CrossRef]
- Bulkin, B.J.; Krishnamachari, N. Vibrational spectra of liquid crystals. IV. Infrared and Raman spectra of phospholipid-water mixtures. J. Am. Chem. Soc. 1972, 94, 1109–1112. [Google Scholar] [CrossRef]
- Bulkin, B.J.; Krishnan, K. Vibrational spectra of liquid crystals. III. Raman spectra of crystal, cholesteric, and isotropic cholesterol esters, 2800-3100-cm-1region. J. Am. Chem. Soc. 1971, 93, 5998–6004. [Google Scholar] [CrossRef]
- Larsson, K. Conformation-dependent features in the raman spectra of simple lipids. Chem. Phys. Lipids 1973, 10, 165–176. [Google Scholar] [CrossRef]
- Larsson, K.; Rand, R.P. Detection of changes in the environment of hydrocarbon chains by raman spectroscopy and its application to lipid-protein systems. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1973, 326, 245–255. [Google Scholar] [CrossRef]
- Gaber, B.P.; Peticolas, W.L. On the quantitative interpretation of biomembrane structure by Raman spectroscopy. Biochim. Biophys. Acta BBA Biomembr. 1977, 465, 260–274. [Google Scholar] [CrossRef]
- Snyder, R.G.; Hsu, S.L.; Krimm, S. Vibrational spectra in the C-H stretching region and the structure of the polymethylene chain. Spectrochim. Acta Part Mol. Spectrosc. 1978, 34, 395–406. [Google Scholar] [CrossRef]
- Snyder, R.G.; Scherer, J.R. Band structure in the C-H stretching region of the Raman spectrum of the extended polymethylene chain: Influence of Fermi resonance. J. Chem. Phys. 1979, 71, 3221–3228. [Google Scholar] [CrossRef]
- Nakamizo, M.; Kammereck, R.; Walker, P.L. Laser raman studies on carbons. Carbon 1974, 12, 259–267. [Google Scholar] [CrossRef]
- Kitson, R.E.; Griffith, N.E. Infrared Absorption Band Due to Nitrile Stretching Vibration. Anal. Chem. 1952, 24, 334–337. [Google Scholar] [CrossRef]
- Ulrich, H. Cumulenes in Click Reactions; Wiley-Interscience: Chichester, UK, 2009. [Google Scholar]
- Sum, A.K.; Burruss, R.C.; Sloan, E.D. Measurement of Clathrate Hydrates via Raman Spectroscopy. J. Phys. Chem. B 1997, 101, 7371–7377. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, W.E.; Cui, L.; Wagner, M. Single cell stable isotope probing in microbiology using Raman microspectroscopy. Curr. Opin. Biotechnol. 2016, 41, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Yu, Y.; Shen, Y.; Wang, M.C.; Min, W. Vibrational imaging of newly synthesized proteins in live cells by stimulated Raman scattering microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 11226–11231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubryk, P.; Kölschbach, J.S.; Marozava, S.; Lueders, T.; Meckenstock, R.U.; Niessner, R.; Ivleva, N.P. Exploring the Potential of Stable Isotope (Resonance) Raman Microspectroscopy and Surface-Enhanced Raman Scattering for the Analysis of Microorganisms at Single Cell Level. Anal. Chem. 2015, 87, 6622–6630. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Mader, E.; Lee, T.K.; Woebken, D.; Wang, Y.; Zhu, D.; Palatinszky, M.; Schintlmeister, A.; Schmid, M.C.; Hanson, B.T.; et al. Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells. Proc. Natl. Acad. Sci. USA 2015, 112, E194–E203. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Corcelli, S.A. Carbon−Deuterium Vibrational Probes of the Protonation State of Histidine in the Gas-Phase and in Aqueous Solution. J. Phys. Chem. B 2010, 114, 8565–8573. [Google Scholar] [CrossRef] [PubMed]
- Frohm, B.; DeNizio, J.E.; Lee, D.S.; Gentile, L.; Olsson, U.; Malm, J.; Åkerfeldt, K.S.; Linse, S. A peptide from human semenogelin I self-assembles into a pH-responsive hydrogel. Soft Matter 2015, 11, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Dasgupta, A.; Mondal, J.H.; Das, D. Peptide hydrogels. RSC Adv. 2013, 3, 9117–9149. [Google Scholar] [CrossRef]
- Hoffman, K.W.; Romei, M.G.; Londergan, C.H. A New Raman Spectroscopic Probe of Both the Protonation State and Noncovalent Interactions of Histidine Residues. J. Phys. Chem. A 2013, 117, 5987–5996. [Google Scholar] [CrossRef]
- Liu, T.; Ryan, M.; Dahlquist, F.W.; Griffith, O. Hayes Determination of pKa values of the histidine side chains of phosphatidylinositol-specific phospholipase C from Bacillus cereus by NMR spectroscopy and site-directed mutagenesis. Protein Sci. 2008, 6, 1937–1944. [Google Scholar] [CrossRef]
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proc. Natl. Acad. Sci. USA 2014, 111, E1705–E1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartik, K.; Redfield, C.; Dobson, C.M. Measurement of the individual pKa values of acidic residues of hen and turkey lysozymes by two-dimensional 1H NMR. Biophys. J. 1994, 66, 1180–1184. [Google Scholar] [CrossRef]
- Bundi, A.; Wüthrich, K. 1H-nmr parameters of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers 1979, 18, 285–297. [Google Scholar] [CrossRef]
- Frassineti, C.; Ghelli, S.; Gans, P.; Sabatini, A.; Moruzzi, M.S.; Vacca, A. Nuclear Magnetic Resonance as a Tool for Determining Protonation Constants of Natural Polyprotic Bases in Solution. Anal. Biochem. 1995, 231, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L. Observation of histidine residues in proteins by nuclear magnetic resonance spectroscopy. Acc. Chem. Res. 1975, 8, 70–80. [Google Scholar] [CrossRef]
- Sachs, D.H.; Schechter, A.N.; Cohen, J.S. Nuclear Magnetic Resonance Titration Curves of Histidine Ring Protons, I. Influence of Neighboring Charged Groups. J. Biol. Chem. 1971, 246, 6576–6580. [Google Scholar] [PubMed]
- Henchoz, Y.; Schappler, J.; Geiser, L.; Prat, J.; Carrupt, P.A.; Veuthey, J.L. Rapid determination of pKa values of 20 amino acids by CZE with UV and capacitively coupled contactless conductivity detections. Anal. Bioanal. Chem. 2007, 389, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Včeláková, K.; Zusková, I.; Kenndler, E.; Gaš, B. Determination of cationic mobilities and pKa values of 22 amino acids by capillary zone electrophoresis. Electrophoresis 2004, 25, 309–317. [Google Scholar] [CrossRef]
- Zscherp, C.; Schlesinger, R.; Tittor, J.; Oesterhelt, D.; Heberle, J. In situ determination of transient pKa changes of internal amino acids of bacteriorhodopsin by using time-resolved attenuated total reflection Fourier-transform infrared spectroscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 5498–5503. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Ono, T.; Noguchi, T. Vibrational Spectra and Ab Initio DFT Calculations of 4-Methylimidazole and Its Different Protonation Forms: Infrared and Raman Markers of the Protonation State of a Histidine Side Chain. J. Phys. Chem. B 2000, 104, 4253–4265. [Google Scholar] [CrossRef]
- Ashikawa, I.; Itoh, K. Raman spectra of polypeptides containing L-histidine residues and tautomerism of imidazole side chain. Biopolymers 1979, 18, 1859–1876. [Google Scholar] [CrossRef]
- Walba, H.; Isensee, R.W. Acidity Constants of Some Arylimidazoles and Their Cations. J. Org. Chem. 1961, 26, 2789–2791. [Google Scholar] [CrossRef]
- Kumar, R.; Das, S.; Mohite, G.M.; Rout, S.K.; Halder, S.; Jha, N.N.; Ray, S.; Mehra, S.; Agarwal, V.; Maji, S.K. Cytotoxic Oligomers and Fibrils Trapped in a Gel-like State of α-Synuclein Assemblies. Angew. Chem. Int. Ed. 2018, 57, 5262–5266. [Google Scholar] [CrossRef] [PubMed]
- Semerdzhiev, S.A.; Shvadchak, V.V.; Subramaniam, V.; Claessens, M.M.A.E. Non-uniform self-assembly: On the anisotropic architecture of α-synuclein supra-fibrillar aggregates. Sci. Rep. 2017, 7, 7699. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Foley, A.C.; Vaux, D.J.T. The Physiological and Pathological Implications of the Formation of Hydrogels, with a Specific Focus on Amyloid Polypeptides. Biomolecules 2017, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Altunbas, A.; Lee, S.J.; Rajasekaran, S.A.; Schneider, J.P.; Pochan, D.J. Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials 2011, 32, 5906–5914. [Google Scholar] [CrossRef] [Green Version]
- Alix, A.J.P.; Pedanou, G.; Berjot, M. Fast determination of the quantitative secondary structure of proteins by using some parameters of the Raman Amide I band. J. Mol. Struct. 1988, 174, 159–164. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pogostin, B.H.; Malmendal, A.; Londergan, C.H.; Åkerfeldt, K.S. pKa Determination of a Histidine Residue in a Short Peptide Using Raman Spectroscopy. Molecules 2019, 24, 405. https://doi.org/10.3390/molecules24030405
Pogostin BH, Malmendal A, Londergan CH, Åkerfeldt KS. pKa Determination of a Histidine Residue in a Short Peptide Using Raman Spectroscopy. Molecules. 2019; 24(3):405. https://doi.org/10.3390/molecules24030405
Chicago/Turabian StylePogostin, Brett H., Anders Malmendal, Casey H. Londergan, and Karin S. Åkerfeldt. 2019. "pKa Determination of a Histidine Residue in a Short Peptide Using Raman Spectroscopy" Molecules 24, no. 3: 405. https://doi.org/10.3390/molecules24030405
APA StylePogostin, B. H., Malmendal, A., Londergan, C. H., & Åkerfeldt, K. S. (2019). pKa Determination of a Histidine Residue in a Short Peptide Using Raman Spectroscopy. Molecules, 24(3), 405. https://doi.org/10.3390/molecules24030405