Polar Metallocenes


Abstract
:
1. Introduction
2. Assessing van der Waals (vdW) Functionals
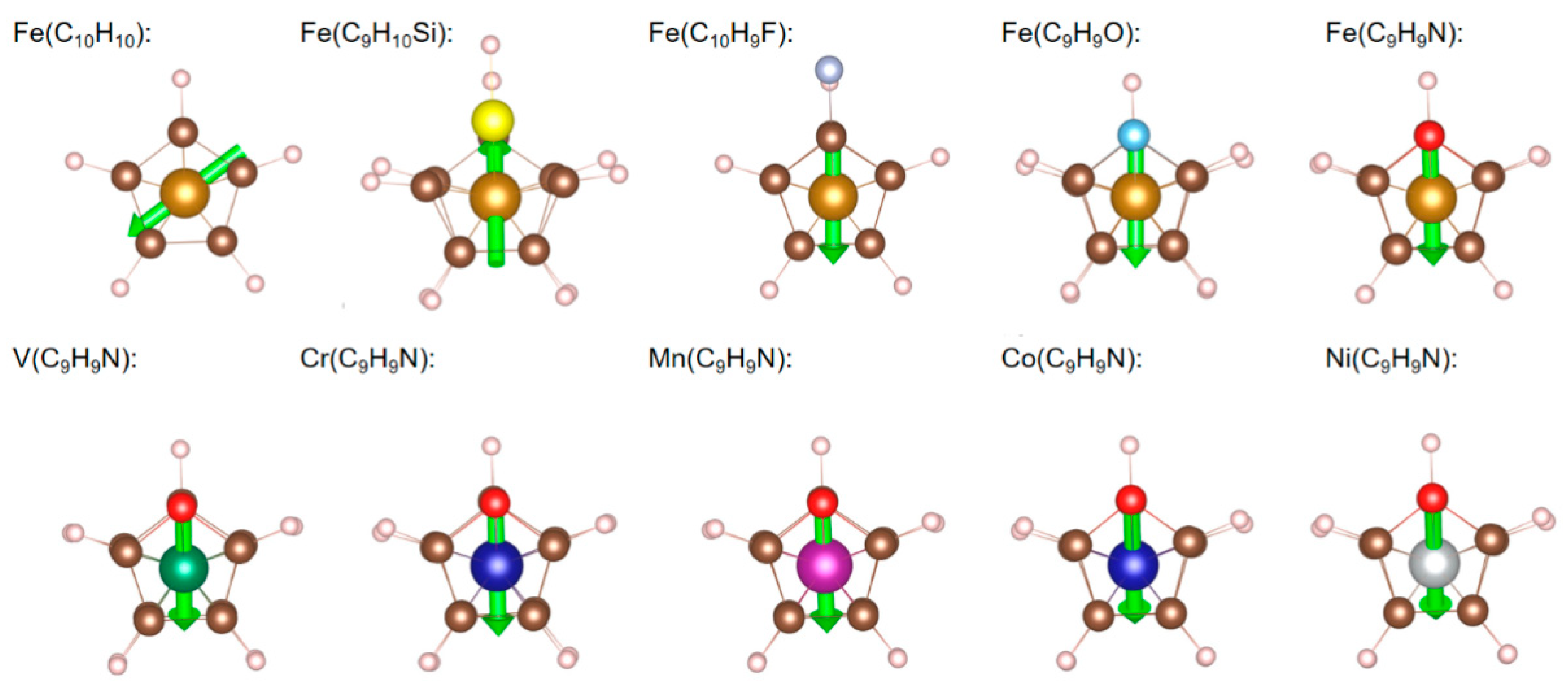
3. Dipole Moments for Isolated Molecules
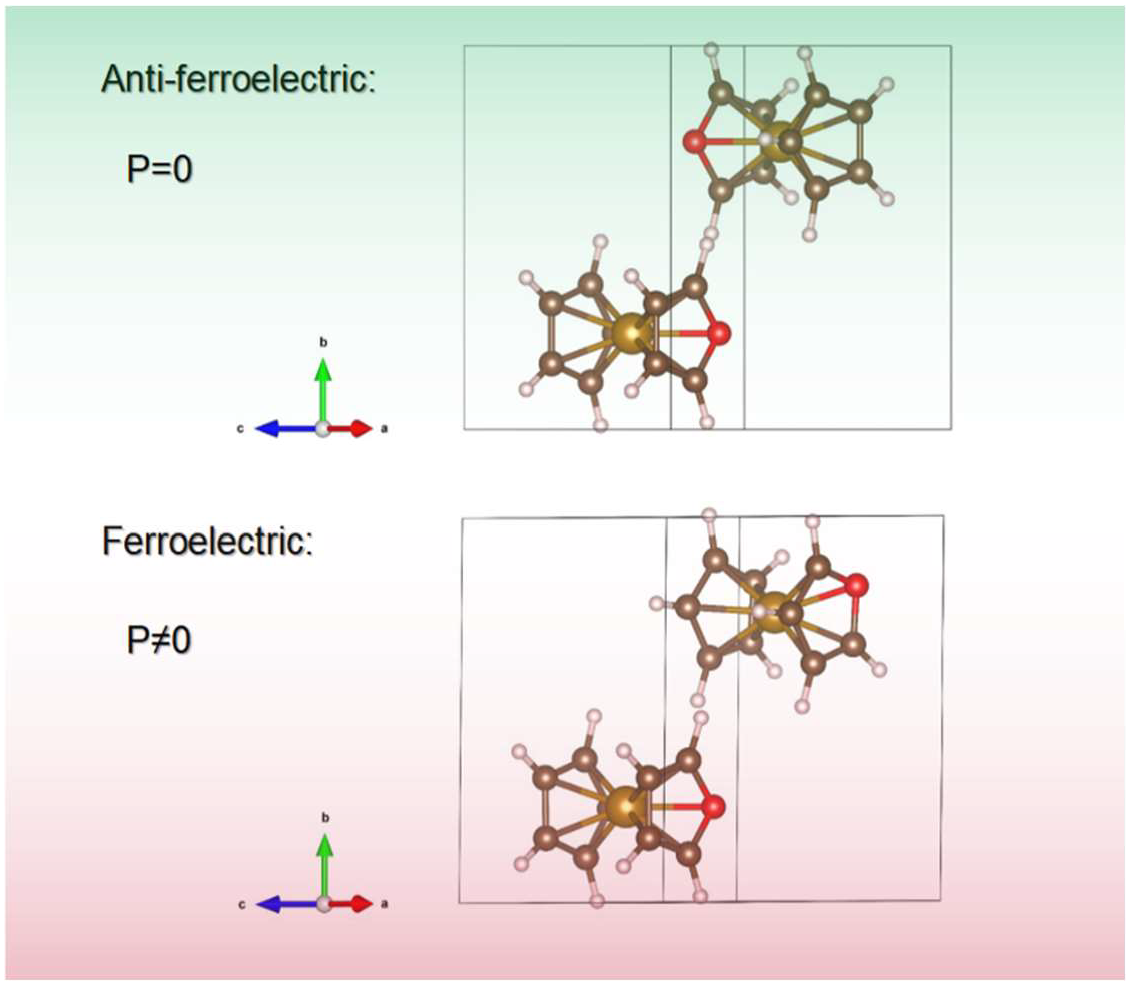
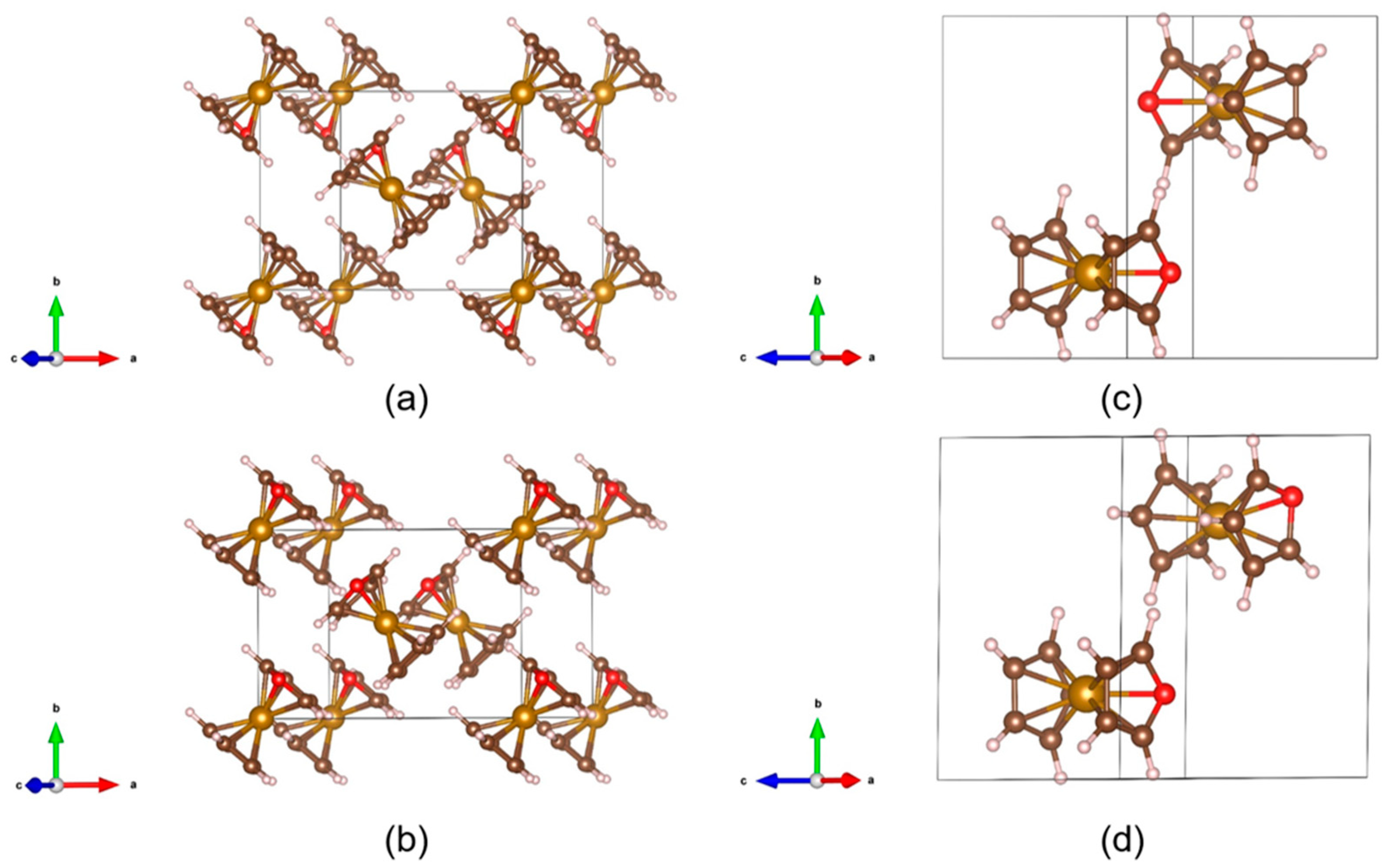
4. Crystalline Polar Metallocenes
5. Materials and Methods
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicolaev, G.A.; Nicolaev, A. Transport in ferrocene single molecules for terahertz applications. Phys. Chem. Chem. Phys. 2014, 16, 5. [Google Scholar]
- Cunha, T.F.D.; Calderini, D.; Skouteris, D. Analysis of Partition Functions for Metallocenes: Ferrocene, Ruthenocene, and Osmocene. J. Phys. Chem. A. 2016, 120, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Jiang, X.; Hu, J.; Huang, X.; Zhao, J.; Ahuja, R. Prediction of huge magnetic anisotropies in 5d transition metallocenes. J. Phys. Condens. Matter. 2017, 29, 435802. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.; Pfab, W. Cyclopentadien-Metallkomplexe, ein neuer Typ metallorganischer Verbindungen. Z. Nat. B 1952, 7, 3. [Google Scholar] [CrossRef]
- Wilkinson, G.R.M.; Whiting, M.C. The Structure of Iron Bis-Cyclopentadienyl. J. Am. Chem. Soc. 1952, 74, 2125–2126. [Google Scholar] [CrossRef]
- Frunzke, J.; Lein, M.; Frenking, G. Structures, Metal-Ligand Bond Strength, and Bonding Analysis of Ferrocene Derivatives with Group-15 Heteroligands Fe(η5-E5)2 and FeCp(η5-E5) (E=N, P, As, Sb). A Theoretical Study. Organometallics 2002, 21, 3351–3359. [Google Scholar] [CrossRef]
- Dombrowski, K.E.; Baldwin, W.; Sheats, J.E. Metallocenes in biochemistry, microbiology and medicine. J. Organomet. Chem. 1986, 302, 26. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Orgel, L.E. Electronic Structure of Metal biscyclopentadienyls. J. Chem. Phys. 1955, 23, 954–958. [Google Scholar] [CrossRef]
- Vargas-Caamal, A.; Pan, S.; Ortiz-Chi, F.; Cabellos, J.L.; Boto, R.A.; Contreras-Garcia, J.; Restrepo, A.; Chattaraj, P.K.; Merino, G. How strong are the metallocene–metallocene interactions? Cases of ferrocene, ruthenocene, and osmocene. Phys. Chem. Chem. Phys. 2016, 18, 550–556. [Google Scholar] [CrossRef]
- Sünkel, K.; Weigand, S.; Hoffmann, A.; Blomeyer, S.; Reuter, C.G.; Vishnevskiy, Y.V.; Mitzel, N.W. Synthesis and Characterization of 1,2,3,4,5-Pentafluoroferrocene. J. Am. Chem. Soc. 2015, 137, 126–129. [Google Scholar] [CrossRef]
- Sohn, Y.S.; Hendrickson, D.N.; Gray, H.B. Electronic Structure of Metallocenes. J. Am. Chem. Soc. 1971, 93, 3603–3612. [Google Scholar]
- Haaland, A. Molecular Structure and Bonding in the 3d Metallocenes. Acc. Chem. Res. 1979, 12, 415–422. [Google Scholar] [CrossRef]
- Little, W.F. Metallocenes. Surv. Prog. Chem. 1963, 1, 78. [Google Scholar]
- Wu, X.; Zeng, X. Double Metallocene Nanowires. J. Am. Chem. Soc. 2009, 131, 14246–14248. [Google Scholar] [CrossRef] [PubMed]
- Seiler, P.; Dunitz, J.D. A New Interpretation of the Disordered Crystal Structure of Ferrocene. Acta Cryst. 1979, 35, 1068–1074. [Google Scholar] [CrossRef]
- Dunitz, J.D. Phase Changes and Chemical Reactions in Molecular Crystals. Acta Cryst. B 1995, 51, 619–631. [Google Scholar] [CrossRef]
- Seiler, P.; Dunitz, J.D. Low-temperature Crystallization of Orthorhombic Ferrocene: Structure Analysis at 98 K. Acta Cryst. Sect. B-Struct. Sci. 1982, 38, 1741–1745. [Google Scholar] [CrossRef]
- Seiler, P.; Dunitz, J.D. The Structure of Triclinic Ferrocene at 101, 123 and 148 K. Acta Cryst. 1979, B35, 2020–2032. [Google Scholar] [CrossRef]
- Edwards, J.W.; Kington, G.L.; Mason, R. The thermodynamic properties of ferrocene. Trans. Faraday Soc. 1960, 56, 660–667. [Google Scholar] [CrossRef]
- Bermúdez-García, J.M.; Yáñez-Vilar, S.; Castro-García, S.; Señarís-Rodríguez, M.A.; Sánchez-Andújar, M. New properties in old systems: Cooperative electric order in ferrocene and ammonia-borane. RSC Adv. 2015, 5, 83818–83824. [Google Scholar] [CrossRef]
- Wu, M.; Burton, J.D.; Tsymbal, E.Y.; Zeng, X.C.; Jena, P. Multiferroic Materials Based on Organic Transition-Metal Molecular Nanowires. J. Am. Chem. Soc. 2012, 134, 14423–14429. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Yang, S.-W.; Ng, M.-F.; Ligatchev, V.; Zhou, L.; Feng, Y. Charge-Transfer-Based Mechanism for Half-Metallicity and Ferromagnetism in One-Dimensional Organometallic Sandwich Molecular Wires. J. Am. Chem. Soc. 2008, 130, 13956–13960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Gao, Y.; Zeng, X. Ab Initio Study of Structural and Magnetic Properties of TMn(ferrocene)n+1 (TM = Sc, Ti, V, Mn) Sandwich Clusters and Nanowires (n = ∞). ACS Nano 2009, 3, 537–545. [Google Scholar] [CrossRef]
- Fey, N. Organometallic molecular modelling-the computational chemistry of metallocenes: A review. J. Chem. Technol. Biotechnol. 1999, 74, 852–862. [Google Scholar] [CrossRef]
- Coriani, S.; Haaland, A.; Helgaker, T.; Jørgensen, P. The Equilibrium Structure of Ferrocene. ChemPhysChem 2006, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Aliabad, H.A.R.; Chahkandi, M. Comprehensive SPHYB and B3LYP-DFT Studies of Two Types of Ferrocene. Z. Anorg. Allg. Chem 2017, 643, 420–431. [Google Scholar] [CrossRef]
- Xu, Z.-F.; Xie, Y.; Feng, W.-L.; Schaefer, H.F. Systematic Investigation of Electronic and Molecular Structures for the First Transition Metal Series Metallocenes M(C5H5)2 (M=V, Cr, Mn, Fe, Co, and Ni). J. Phys. Chem. A 2003, 107, 2716–2729. [Google Scholar] [CrossRef]
- Seller, P.; Duntitz, J.D. The Structure of Nickelocene at Room Temperature and at 101 K. Acta Cryst. 1980, 36, 2255–2260. [Google Scholar] [CrossRef]
- Yu, M.; Antipin, R.B. Redetermination of the Cobaltocene Crystal Structure at 100 K and 297 K: Comparison with Ferrocene and Nickelocene. Struct. Chem. 1993, 4, 91–101. [Google Scholar]
- Yu, M.; Antipin, R.B. Structure of Vanadocene in the Temperature Interval 108–357 K and the Nature of the Ring Disorder. Acta Cryst. 1996, 52, 314–322. [Google Scholar]
- Robertson, R.E.; Mcconnell, H.M. The magnetic resonance properties of some sandwich compounds. J. Phys. Chem. 1960, 64, 70–77. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Marzari, N.; Mostofi, A.A.; Yates, J.R.; Souza, I.; Vanderbilt, D. Maximally localized Wannier functions: Theory and applications. Rev. Mod. Phys. 2012, 84, 1419–1475. [Google Scholar] [CrossRef]
- Rosenblum, M.; Fish, R.W. A convenient synthesis of some haloferrocenes. J. Org. Chem. 1965, 30, 1253–1254. [Google Scholar]
- Bernhartzeder, S.; Sunkel, K. Coordination chemistry of perhalogenated cyclopentadienes and alkynes. Part 30. New high-yield syntheses of monochloroferrocene and 1,2,3,4,5-pentachloroferrocene. Molecular structures of 1,2-dichloroferrocene and 1,2,3-trichloroferrocene. J. Organomet. Chem. 2012, 716, 146–149. [Google Scholar] [CrossRef]
- Sunkel, K.; Bernhartzeder, S. Coordination chemistry of perhalogenated cyclopentadienes and alkynes. XXVIII [1] new high-yield synthesis of monobromoferrocene and simplified procedure for the synthesis of pentabromoferrocene. Molecular structures of 1,2,3-tribromoferrocene and 1,2,3,4,5-pentabromoferrocene. J. Organomet. Chem. 2011, 696, 1536–1540. [Google Scholar]
- Sünkel, K. Coordination chemistry of pentahalocyclopentadienyls. Chem. Ber. -Recl. 1997, 130, 1721–1730. [Google Scholar]
- Hnetinka, C.A.; Hunter, A.D.; Zeller, M.; Lesley, M.J.G. 1,1′-Dibromoferrocene. Acta Cryst. E 2004, 60, m1806–m1807. [Google Scholar] [CrossRef]
- Romanov, A.S.; Mulroy, J.M.; Khrustalev, V.N.; Antipin, M.Y.; Timofeeva, T.V. Monohalogenated ferrocenes C5H5FeC5H4X (X = Cl, Br and I) and a second polymorph of C5H5FeC5H4I. Acta Cryst. C 2009, 65, m426–m430. [Google Scholar] [CrossRef]
- Chaudret, B.; Jalon, F.A. Facile preparation of π-arene complexes of ruthenium [(η5-C5Me5)Ru(Arene)]X including a π-pyridine and the first π-furan derivatives. J. Chem. Soc. Chem. Commun. 1988, 711–713. [Google Scholar] [CrossRef]
- Da, H.; Jin, H.M.; Lim, K.H.; Ligatchev, V.; Ng, M.-F.; Khoo, K.H.; Yang, S.-W. Half metal semiconductor reversible switch in bimetallic sandwich molecular wire via redox reactions. Nano Energy 2012, 1, 297–302. [Google Scholar] [CrossRef]
- Gardner, A.B.; Howard, J.; Waddington, T.C. The dynamics of ring rotation in ferrocene, nickelocene and ruthenocene by incoherent quasi-elastic neutron scattering. Chem. Phys. 1981, 57, 453–460. [Google Scholar] [CrossRef]
- Brock, C.P.; Fu, Y. Rigid-Body Disorder Models for the High-Temperature Phase of Ferrocene. Acta Cryst. 1997, 928–938. [Google Scholar] [CrossRef]
- Clec’h, G.; Calvarin, G. Etude Structurale Des Phases Desordonnees De Co(C5H5)2, Mg(C5H5)2, Et Transitions De Phases De (C4H4)N)Fe(C5H5). Mol. Cryst. Liq. Cryst. 2011, 128, 305–320. [Google Scholar] [CrossRef]
- Chhor, K.; Pommier, C. Low temperature thermodynamic study of the stable and metastable phases of (C4H4N)Fe(C5H5). J. Chem. Thermodyn. 1984, 16, 503–517. [Google Scholar] [CrossRef]
- Gregory, J.K.; Clary, D.C.; Liu, K.; Brown, M.G.; Saykally, R.J. The Water Dipole Moment in Water Clusters. Science. 1997, 275, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Silvestrelli, P.L.; Parrinello, M. Water Molecule Dipole in the Gas and in the Liquid Phase. Phys. Rev. Lett. 1999, 82, 3308–3311. [Google Scholar] [CrossRef]
- Shieh, J.; Yeh, J.H.; Shu, Y.C.; Yen, J.H. Hysteresis behaviors of barium titanate single crystals based on the operation of multiple 90° switching systems. Mater. Sci. Eng. B 2009, 161, 50–54. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Sabatini, R.; Gorni, T.; de Gironcoli, S. Nonlocal van der Waals density functional made simple and efficient. Phys. Rev. B. 2013, 87, 4. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Van Voorhis, T. Nonlocal van der Waals density functional: The simpler the better. J. Chem. Phys. 2010, 133, 10. [Google Scholar] [CrossRef] [PubMed]
- Thonhauser, T.; Zuluaga, S.; Arter, C.A.; Berland, K.; Schroder, E.; Hyldgaard, P. Spin Signature of Nonlocal Correlation Binding in Metal-Organic Frameworks. Phys. Rev. Lett. 2015, 115, 136402. [Google Scholar] [CrossRef] [PubMed]
- Thonhauser, T.; Cooper, V.R.; Li, S.; Puzder, A.; Hyldgaard, P.; Langreth, D.C. Van der Waals density functional: Self-consistent potential and the nature of the van der Waals bond. Phys. Rev. B 2007, 76, 11. [Google Scholar] [CrossRef]
- Langreth, D.C.; Dion, M.; Rydberg, H.; Schröder, E.; Hyldgaard, P.; Lundqvist, B.I. Van der Waals density functional theory with applications. Int. J. Quantum Chem. 2005, 101, 599–610. [Google Scholar] [CrossRef]
- Murray, E.D.; Lee, K.; Langreth, D.C. Investigation of Exchange Energy Density Functional Accuracy for Interacting Molecules. J. Chem. Theory Comput. 2009, 5, 2754–2762. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Murray, E.D.; Kong, L.; Lundquist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B. 2010, 82, 081101(R). [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| DFT-D | rVV10 | vdW-DF2 | Exp | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FM | AFM | NM | FM | AFM | NM | FM | AFM | NM | |||
| Fe2(C10H10)2 | ΔEMS | 2.222 | - | 0.00 | 1.871 | - | 0.00 | 1.55 | - | 0.00 | - |
| M | 4.05 | - | 0.00 | 4.05 | - | 0.00 | 4.05 | - | 0.0 | 0.00 a | |
| Vcell | 366.43 | - | 344.37 | 387.12 | - | 363.45 | 416.37 | - | 391.85 | 395.00 b | |
| Ni2(C10H10)2 | ΔEMS | 0.0 | 0.001 | 0.310 | 0.00 | - | 0.365 | 0.00 | 0.037 | 0.166 | - |
| M | 2.18 | 2.15 | 0.00 | 2.18 | - | 0.00 | 2.17 | 2.15 | 0.00 | 2.89 a | |
| Vcell | 358.10 | 358.23 | 353.87 | 373.25 | - | 370.79 | 398.08 | 384.73 | 412.09 | 403.67 b | |
| Co2(C10H10)2 | ΔEMS | 1.181 | 0.00 | 0.225 | 0.948 | 0.00 | 1.432 | 0.755 | 0.00 | 0.468 | - |
| M | 3.11 | 1.24 | 0.00 | 3.11 | 1.25 | 0.00 | 3.10 | 1.25 | 0.00 | 1.73 a | |
| Vcell | 366.09 | 351.22 | 350.59 | 386.45 | 371.93 | 450.32 | 421.09 | 405.40 | 462.36 | 418.57 b | |
| V2(C10H10)2 | ΔEMS | 0.003 | 0.00 | 1.200 | 0.003 | 0.00 | 1.252 | 0.001 | 0.000 | 1.104 | - |
| M | 3.11 | 3.11 | 0.00 | 3.09 | 3.18 | 0.00 | 3.16 | 3.08 | 0.00 | 3.78 a | |
| Vcell | 370.57 | 369.95 | 356.99 | 387.93 | 387.48 | 376.42 | 418.79 | 418.54 | 418.97 | 414.77 b | |
| Fe(C10H10) | Fe(C9SiH10) | Fe(C10H9F) | Fe(C9H9O) | Fe(C9H9N) | V(C9H9N) | Cr(C9H9N) | Mn(C9H9N) | Co(C9H9N) | Ni(C9H9N) | |
|---|---|---|---|---|---|---|---|---|---|---|
| N | 6 | 6 | 6 | 7 | 6 | 3 | 4 | 5 | 7 | 8 |
| M | 0 | 0 | 0 | 1 | 0 | 3 | 2 | 1 | 1 | 2 |
| μ | 0.08 | 0.25 | 1.70 | 1.32 | 2.08 | 1.75 | 2.37 | 1.84 | 2.20 | 2.80 |
| Erotation | 2.61 | 3.38 | 2.41 | −3.76 | 2.22 | 0.29 | 0.97 | 2.12 | 1.06 | −1.74 |
| Δμ | −0.01 | 0.04 | 0.04 | 0.07 | −0.25 | 0.31 | −0.40 | 0.26 | 0.02 | −0.03 |
| Fe(C10H9Cl) | Fe(C10H9Br) | Fe(C10H9I) | |
|---|---|---|---|
| μ | 1.77 | 1.82 | 1.81 |
| Eact | 2.19 | 2.16 | 2.01 |
| Δμ | 0.01 | −0.21 | −0.02 |
| Fe(C9H9B) | Fe(C9H9P) | Fe(C9H9As) | Fe(C9H9Sb) | Fe(C9H9Bi) | |
|---|---|---|---|---|---|
| μ | 0.87 | 1.71 | 1.42 | 0.70 | 0.52 |
| Eact | 3.41 | 2.60 | 3.60 | 3.08 | 3.19 |
| Δμ | 0.10 | 0.01 | −0.11 | 0.16 | 0.30 |
| Fe2(C10H9F)2 | Fe2(C9H9O)2 | V2(C9H9N)2 | Cr2(C9H9N)2 | Mn2(C9H9N)2 | Fe2(C9H9N)2 | Co2(C9H9N)2 | Ni2(C9H9N)2 | |
|---|---|---|---|---|---|---|---|---|
| Vcell | 404.58 | 382.58 | 404.87 | 392.73 | 386.79 | 378.17 | 385.17 | 382.98 |
| ΔEpolar | 80.96 | 74.96 | 12.22 | 5.99 | −0.87 | 17.67 | 44.78 | 6.53 |
| Ebind | 0.89 | 0.76 | 0.90 | 0.85 | 0.76 | 0.91 | 0.87 | 0.57 |
| Fe2(C10H9F)2 | Fe2(C9H9O)2 | V2(C9H9N)2 | Cr2(C9H9N)2 | Mn2(C9H9N)2 | Fe2(C9H9N)2 | Co2(C9H9N)2 | Ni2(C9H9N)2 | |
|---|---|---|---|---|---|---|---|---|
| MGS | / | AFM | AFM | AFM | FM | / | FM | AFM |
| ΔEMS | / | −7.92 | −6.20 | −1.60 | 0.47 | / | 47.57 | −4.93 |
| Mcell | 0 | 0 | 0 | 0 | 2.0 | 0 | 2.0 | 0 |
| P | 4.40 | 3.16 | 5.03 | 6.06 | 5.43 | 5.56 | 5.79 | 6.56 |
| P* | 2.72 | 1.91 | 2.77 | 3.92 | 3.07 | 3.55 | 3.69 | 4.80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Yavorsky, B.Y.; Cohen, R.E. Polar Metallocenes. Molecules 2019, 24, 486. https://doi.org/10.3390/molecules24030486
Zhang H, Yavorsky BY, Cohen RE. Polar Metallocenes. Molecules. 2019; 24(3):486. https://doi.org/10.3390/molecules24030486
Chicago/Turabian StyleZhang, Haiwu, B.Yu. Yavorsky, and R.E. Cohen. 2019. "Polar Metallocenes" Molecules 24, no. 3: 486. https://doi.org/10.3390/molecules24030486
APA StyleZhang, H., Yavorsky, B. Y., & Cohen, R. E. (2019). Polar Metallocenes. Molecules, 24(3), 486. https://doi.org/10.3390/molecules24030486