1. Introduction
Maclura tricuspidata (Carr.) Bur (formerly known as
Cudrania tricuspidata), which belongs to the Moraceae family, is a thorny tree native to East Asia, including China, Japan, and Korea. The leaves, root, stem, and fruit of this plant have been used in traditional Korean herbal medicines to treat jaundice, hepatitis, neuritis, and inflammation [
1]. Over the last two decades, several beneficial effects of
M. tricuspidata extracts have been reported; they include anticancer [
2,
3], anti-inflammatory [
4], antioxidant [
5,
6], oxidative stress-induced neurotoxicity [
7], anti-obesity and anti-diabetes properties [
8]. Moreover, a variety of bioactive compounds such as prenylated xanthones, phenolic acids, and flavonoids have already been identified from its leaves, root, stem, and fruit [
9,
10,
11]. These compounds were also reported to have antitumor [
11], antibacterial [
12], antioxidant [
13,
14], neuroprotective [
1], cytotoxic [
13,
15], anti-inflammatory [
16], hepatoprotective [
17], gastroprotective [
16], and α-glucosidase inhibition activities [
18].
Polyphenols in a plant diet gained much interest mainly due to their antioxidant properties, which provide health benefits such as anti-inflammatory, anti-carcinogenic, anti-atherogenic, antithrombotic, immune-enhancement, and vasodilatory effects [
19]. These compounds act as scavengers of reactive oxygen species such as hydroxyl, hydroperoxyl, and superoxide anion radicals, which are inevitably produced by the metabolic reactions of living systems [
20,
21]. These radical species attack lipids, proteins and DNA, damaging biological structures, including cell membranes, enzymes, and genetic material. This damage can cause various chronic diseases such as cancer, cardiovascular diseases, atherosclerosis, hypertension, diabetes, neurodegenerative disorders, rheumatoid arthritis, and aging [
22].
Both the premature and fully mature fruits of M. tricuspidata have traditionally been used in Korea to make fresh juice, jam, wine, vinegar, and fermented alcoholic beverages. The cultivation area of this plant for fruit production has recently increased substantively and is to some extent being popularized. While interest in its extracts has steadily increased due to its diverse biological activities, little is known about the bioactive compounds of its fruit when compared to those of its leaves, root, and bark. Due to this, there is limited information available about variations in the level of total polyphenols, individual polyphenols, and antioxidant activities of the fruit at different maturity stages.
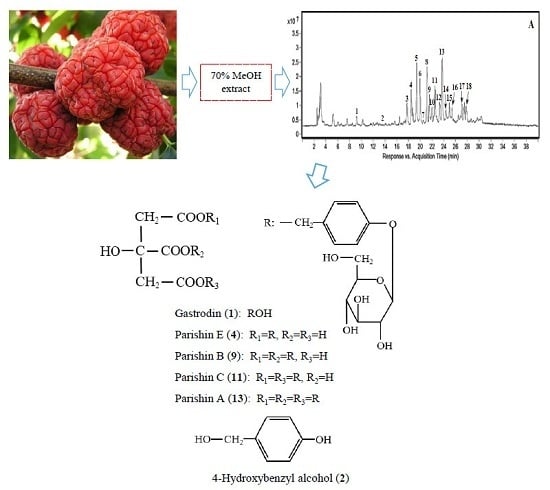
This study aimed at exploring the variations of total polyphenols, individual polyphenolic compounds, and antioxidant activities at different maturity stages of the M. tricuspidata fruit. We developed systematic methods of HPLC/Q-TOF-MS and reverse phase high performance liquid chromatography (RP-HPLC) with photo-diode array detection (DAD) to identify and quantify newly discovered compounds. We were able to identify five parishin derivatives (gastrodin, parishin A, B, C, E) for the first time.
3. Materials and Methods
3.1. Reagents
Chlorogenic acid, taxifolin, eriodictyol, quercetin, rutin, kaempferol, 4-hydroxybenzyl alcohol (4-HBA), β-carotene, 2,2-diphenyl-1-picrylhydrazyl (DPPH), 6-Hydroxy-2,5,7,8-tetramethylchromane- 2-carboxylic acid (Trolox), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), 2,4,6-tri(2-pyridyl)-s-triazine (TPTZ) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Gastrodin, parishin A, parishin B, parishin C and parishin E were purchased from Chengdu Biopurify Phytochemicals Ltd. (Chengdu, Sichuan, China). HPLC grade deionized water, acetone and methanol were purchased from J.T. Baker (Center Valley, PA, USA). The other reagents used were of analytical grade and were purchased from Daihan Scientific Co., Ltd. (Wonju, Korea).
3.2. Plant Materials
Maclura tricuspidata fruit was collected in late October 2015 from plants cultivated in a farm located in Milyang district, Gyeongsangnam-do, Republic of Korea. Voucher specimen was deposited at the Herbarium of Department of Food Science and Technology, College of Agricultural Life Science, Chonbuk National University. Next, the fruits were divided into four different maturity stage groups i.e., immature, premature, fully mature and overmature based on their colors by colorimeter and visual appearance. The color was recorded using a Minolta CM3500d (Minolta Camera Co., Ltd., Osaka, Japan) tristimulus colorimeter. The results were expressed as L*, a* and b* values. The L* value indicates darkness or lightness of color and ranges from black (0) to white (100). The a* and b* values indicate color directions; +a is the red direction (+100), –a is the green direction (−80), +b is the yellow direction (+70) and –b is the blue direction (−50). The sorted fruits were freeze-dried. The samples were powdered and stored in a freezer (−20°C) until use.
3.3. Sample Extraction
The powdered sample (1.0 g) was extracted with 20 mL of 70% aqueous methanol with an ultrasonicator (Hwa Shin Instrument Co., Seoul, Korea) at room temperature for 30 min and centrifuged at 4500 rpm for 15 min. The residue was extracted with 20 mL of 70% aqueous methanol followed by centrifugation as above. The supernatants were combined and evaporated under reduced pressure. The residue was typically dissolved in 10 mL of 70% aqueous methanol and diluted when necessary. In order to examine the effect of extraction solvent system on yields of parishin derivatives, powdered sample (1.0 g) was extracted in the same way as above, except different aqueous methanol concentrations (100, 80, 60, 50, 40, 30, 20 and 0%) were used.
3.4. Chromatographic Analysis
3.4.1. HPLC-QTOF-MS
HPLC-QTOF-MS analysis was performed on an Agilent 1200 HPLC system, including a binary pump, an autosampler, and photodiode array detector (DAD) coupled to an Agilent 6550 QTOF-MS (Agilent Technologies, Palo Alto, CA, USA). HPLC analysis was performed using a ZORBAX Eclipse XDB-C18 (250 mm × 4.6 mm, 5 μm, Agilent Technologies, Palo Alto, CA, USA) at 30 °C. The mobile phase consisted of 0.1% formic acid in distilled water (solvent A), and 0.1% formic acid in methanol (solvent B). The gradient elution program was as follows: a linear gradient elution of 5–15% B from 0 to 10 min, 15–55% B from 10 to 25 min, 55–5% B from 25 to 35 min and maintained 5% B for 5 min at a flow rate of 0.8 mL/min.
The serial parameters for QTOF-MS analysis were set as follows: the nebulizer and auxiliary gas (nitrogen), drying gas (nitrogen) flow rate of 15 L/min; drying gas temperature of 225 °C; nebulizer pressure of 45 psi; sheath gas flow of 11 L/min, sheath gas temperature of 350 °C, capillary voltage of 3500 V, fragmentation voltage of 400 V, and collision energy of 0 V. The full-scan data acquisition was performed from 50 to 1200 m/z with a scan time of 1 s. All mass spectrometry data were recorded in negative ion (ESI) mode. Data was processed on the Agilent MassHunter Workstation data acquisition software (ver. B.02.01) and qualitative analysis software (ver. B. 03.01).
3.4.2. HPLC
HPLC analysis was performed using an HPLC system (Waters, Milford, MA, USA) equipped with a 2690 separation module and Waters 996 DAD with an ZORBAX Eclipse XDB-C18 column (250 mm × 4.6 mm, 5 μm; Agilent Technologies, Inc., Santa Clara, CA, USA). The mobile phase consisted of 0.1% formic acid in ionized water (solvent A) and 0.1% formic acid in methanol (solvent B). The ratio of the mobile phase was maintained at A:B 95:5 (0–5 min), 85:15 (5–10 min), 45:55 (10–25 min), and 95:10 (25–40 min) at a flow rate of 0.8 mL/min. UV–Vis absorption spectra were recorded from 200–400 nm during the HPLC analysis and the quantification of individual compounds were based on peak areas at 220 nm.
3.4.3. Quantification and Method Validation
Among 18 compounds identified in this study, 12 compounds were quantified by an external calibration method using chromatograms measured at 220 nm. Standards with the same structural chromophores were used to quantify the individual polyphenols. Accordingly, taxifolin and eriodictyol were used to quantify taxifolin glucoside (1) and eriodictyol glucoside (6), respectively. Quercetin derivatives (7, 12, 16) were quantified using quercetin and dihydrokamempferol (17) and kampferol glucoside (18) were quantified using kaempferol as a reference.
The method linearity for the quantification of each polyphenol was tested on the basis of a calibration curve, which was processed using a linear regression. The seven-point standard solutions (1.25–640 μg/mL) for polyphenols and 6.25–400 μg/mL for parishin derivatives were prepared from serial dilutions of the stock solution (1000 μg/mL) and were used for calibration according to an external method. The areas of the partially overlapped peaks were automatically calculated by the HPLC integrator. The limit of detection (LOD) and limit of quantitation (LOQ) were calculated by injecting standard solution until the signal-to-noise (S/N) ratio of each compound reached ratios of 3.3:1 and 10:1, respectively [
43]. Intra-day and inter-day assays were used to determine the precision of established chromatographic conditions. The sample was extracted and analyzed in five replicates within 1 day for intra-day precision test. For the inter-day precision test, the sample was extracted and analyzed in duplicates for five consecutive days. Precisions were expressed as the relative standard deviation (RSD).
3.5. Determination of Total Carotenoid
Total carotenoid content was analyzed spectrophotometrically according to a method by Biswas et al. [
44], with some modifications. The powdered sample (1.0 g) was extracted with 10 mL of chilled acetone under sonication for 20 min at room temperature (RT) and centrifuged at 4500 rpm for 15 min at 4 °C. The residue was re-extracted with 10 mL of chilled acetone followed by centrifugation as above. Both of the supernatants were combined and filled up to a total volume of 25 mL with acetone. Absorbance was measured at 470 nm in a UV-Vis spectrophotometer. A calibration curve was plotted using authentic standard β-carotene, and the content of total carotenoid was expressed as mg β-carotene equivalent per 100 g of dried weight (mg β-CAE/100 g dw).
3.6. Total Phenol and Total Flavonoid Content
3.6.1. Total Phenol
Total phenol content of the sample was measured according to a method described by Chandra et al. [
45], with some modifications. Briefly, 70% MeOH extract (20 μL) of each sample was mixed with 50% Folin-Ciocalteu phenol reagent (20 μL) in 96-well plates. After 5 min, 1 N sodium carbonate (20 μL) was added to the mixture and distilled water was added to adjust the final volume to 200 μL. After incubation at room temperature in the dark for 30 min, the absorbance of test sample against a blank was measured at wavelength of 725 nm using a VersaMax ELISA microplate reader (Molecular Devices, LLC, CA, USA). Total phenol content was calculated based on a calibration curve of gallic acid. The result was expressed as mg gallic acid equivalent (mg GAE)/100 g dw.
3.6.2. Total Flavonoid
Total flavonoid content was measured according to a method described by Zhishen et al. [
46], with some modifications. Briefly, 70% MeOH extract (30 μL) of each sample was mixed with 30 μL of 5% sodium nitrite solution. After 5 min of reaction, 300 μL of 5% aluminum chloride was added. Then 200 μL of 1 N NaOH was added 6 min later and the total volume was adjusted to 1 mL with distilled water. The absorbance of test sample against a blank was measured at wavelength of 510 nm with a Shimadzu UV-1601 spectrophotometer (Kyoto, Japan). Flavonoid content was calculated using a calibration curve of quercetin. The result was expressed as mg quercetin equivalent (mg QUE)/100 g dw.
3.7. Antioxidant Activity
3.7.1. 1,1-Diphenyl-2-picrylhydrazyl (DPPH) Fee Radical Scavenging Activity
DPPH radical scavenging activity of the sample was determined according to a method described by Thaipong et al. [
47], with some modifications. The stock solution was freshly prepared by dissolving 24 mg DPPH in methanol (100 mL) and the working solution was prepared by diluting stock solution with methanol to obtain an absorbance of 1.1 ± 0.02 units at 517 nm using the UV–Vis spectrophotometer. The 70% methanol extract (20 μL) of
M. tricuspidata fruit was added to 80 μL working solution and 100 μL Tris–HCl buffer (0.1 M). The mixture was shaken at room temperature in dark conditions for 20 min. Absorbance was measured at wavelength of 517 nm using a microplate reader and the calibration curve was linear between 15–150 μM Trolox. The results are expressed as mM TE/g dw.
3.7.2. 2,2′-Azino-bis (3-ethylbenzothiazoline)-6 sulphonic acid (ABTS) Free Radical Scavenging Activity
ABTS free radical scavenging activity was determined by methods described by Thaipong et al. [
47], with some modifications. Briefly, a mixture of ABTS (7.4 mM) solution and potassium persulfate (2.6 mM) solution in 1:1 ratio was kept at room temperature for 12 h under dark conditions to form ABTS cation. The solution was diluted by adding methanol to obtain an absorbance of 1.1 ± 0.02 at 734 nm using a UV-Vis spectrophotometer. All the required solutions were freshly prepared for each assay. 100 μL of the extract was added to 1400 μL of the diluted ABTS solution and the mixture was incubated at room temperature for 2 h in the dark. After the reaction, its absorbance was measured at wavelength of 734 nm. The calibration curve was linear between 15–105 μM Trolox. Results were expressed as mM TE/g dw.
3.7.3. Ferric Reducing Antioxidant Power (FRAP)
Ferric reducing power was determined using FRAP assay [
48], with some modifications. The FRAP reagent was prepared by mixing 10 volumes of 300 mM acetate buffer (pH 3.6) with 1 volume of 10 mM TPTZ solution in 40 mM HCl and 1 volume of 20 mM ferric chloride solution. The sample extract (75 μL) was added to 1425 μL of FRAP reagent. The reaction mixture was incubated at room temperature for 30 min in dark conditions. Then the absorbance of the samples was measured at 593 nm. The calibration curve was linear between 15–180 μM Trolox. Results were expressed as mM TE/g dw.
3.8. Statistical Analysis
All experiments were performed in triplicate, unless otherwise indicated, and the results were expressed as mean ± standard deviation (SD). The statistical analysis was conducted with SPSS (ver. 10.1) for Windows and a one-way analysis of variance (ANOVA). Duncan’s multiple range tests were carried out to test any significant differences among various fruit maturity stages. Values with p < 0.05 were considered as significantly different.
4. Conclusions
Secondary metabolites in plant foods are associated with various biological activities. In fruits, the composition and contents of such metabolites may vary according to maturity levels. Although fully mature M. tricuspidata fruits are usually preferably edible, total phenol, total flavonoids, individual polyphenolic compounds and antioxidant activities were observed to be higher at immature and premature stages than at fully mature and overmature stages. The present phytochemical study revealed the presence of parishin derivatives such as parishin A, B, C and parishin E, and gastrodin originating from parishin analogues in this fruit for the first time in the plant. The contents of these particular compounds were also higher at immature and premature stages than at fully mature and overmature stages with a clearly noticeable decrease of parishin A observed during the maturation. Based on this, it can be concluded that parishin A and its analogues undergo significant changes during maturation of the fruit. Although M. tricuspidata fruit is commonly used for usual food consumption at fully mature and overmature stages, this study showed that the fruit at immature and premature stages can be considered to be preferable as raw material in the production of functional foods. These findings may add valuable information for further exploration of the fruit in terms of its therapeutic and nutritional qualities at different maturity stages.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}