Synthesis of Aromatic Rings Embedded in Polycyclic Scaffolds by Triyne Cycloaddition: Competition between Carbonylative and Non-Carbonylative Pathways


Abstract
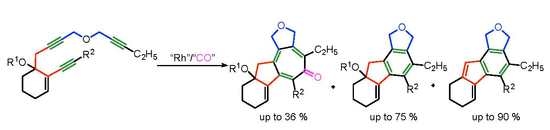
:
1. Introduction
2. Results
2.1. Use of Mo(CO)6 as CO Source for the [2+2+2+1] Carbonylative Cycloaddition
2.2. Use of Carbon Monoxide Gas as CO Source for the [2+2+2+1] Carbonylative Cycloaddition
2.3. Mechanistic Proposals
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef]
- Inglesby, P.A.; Evans, P.A. Higher Order Cycloadditions. In Comprehensive Organic Synthesis II; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 5, pp. 656–704. ISBN 9780080977423. [Google Scholar]
- Domínguez, G.; Pérez-Castells, J. Recent advances in [2+2+2] cycloaddition reactions. Chem. Soc. Rev. 2011, 40, 3430–3444. [Google Scholar] [CrossRef] [PubMed]
- Bennacer, B.; Fujiwara, M.; Ojima, I. Novel [2+2+2+1] cycloaddition of enediynes catalyzed by rhodium complexes. Org. Lett. 2004, 6, 3589–3591. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.W.; Teng, Y.H.G.; Honda, T.; Ojima, I. Synthesis of Colchicinoids and Allocolchicinoids through Rh(I)-Catalyzed [2+2+2+1] and [2+2+2] Cycloadditions of o-Phenylenetriynes with and without CO. J. Org. Chem. 2018, 83, 11623–11644. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, S.I.; Choi, S.Y.; Chung, Y.K. Rhodium-Catalyzed Carbonylative [3+3+1] Cycloaddition of Biscyclopropanes with a Vinyl Substituent To Form Seven-Membered Rings. Angew. Chem. Int. Ed. 2008, 47, 4914–4917. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Gamber, G.G.; Hubbard, R.D.; Zhang, L. Three-Component Cycloadditions: The First Transition Metal-Catalyzed [5+2+1] Cycloaddition Reactions. J. Am. Chem. Soc. 2002, 124, 2876–2877. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Su, J.; Huang, F.; Jiao, L.; Liang, Y.; Yang, D.; Zhang, S.; Wender, P.A.; Yu, Z. A Computationally Designed Rh (I) -Catalyzed Two-Component [5+2+1] Cycloaddition of Ene-vinylcyclopropanes and CO for the Synthesis of Cyclooctenones. J. Am. Chem. Soc. 2007, 129, 10060–10061. [Google Scholar] [CrossRef]
- Brummond, K.M.; Kent, J.L. Recent advances in the Pauson-Khand reaction and related [2+2+1] cycloadditions. Tetrahedron 2000, 56, 3263–3283. [Google Scholar] [CrossRef]
- Salacz, L.; Girard, N.; Suffert, J.; Blond, G. Carbonylative cycloaddition for the synthesis of medium-sized carbo- and heterocycles. Monatshefte Chem. 2018, 149, 671–686. [Google Scholar] [CrossRef]
- Park, J.H.; Chang, K.-M.; Chung, Y.K. Catalytic Pauson–Khand-type reactions and related carbonylative cycloaddition reactions. Coord. Chem. Rev. 2009, 253, 2461–2480. [Google Scholar] [CrossRef]
- Shibata, Y.; Tanaka, K. Rhodium-catalyzed [2+2+2] cycloaddition of alkynes for the synthesis of substituted benzenes: Catalysts, reaction scope, and synthetic applications. Synthesis 2012, 44, 323–350. [Google Scholar] [CrossRef]
- Blouin, S.; Gandon, V.; Blond, G.; Suffert, J. Synthesis of Cyclooctatetraenes through a Palladium-Catalyzed Cascade Reaction. Angew. Chem. Int. Ed. 2016, 55, 7208–7211. [Google Scholar] [CrossRef] [PubMed]
- Joussot, J.; Schoenfelder, A.; Suffert, J.; Blond, G. Synthesis of original polycycles containing five-, six- and seven-membered rings through cyclocarbopalladations/C–H activation cascade reactions. C. R. Chim. 2017, 20, 665–681. [Google Scholar] [CrossRef]
- Petrignet, J.; Boudhar, A.; Blond, G.; Suffert, J. Step-economical synthesis of taxol-like tricycles through a palladium-catalyzed domino reaction. Angew. Chem. Int. Ed. 2011, 50, 3285–3289. [Google Scholar] [CrossRef] [PubMed]
- Charpenay, M.; Boudhar, A.; Blond, G.; Suffert, J. An expeditious and atom-economical synthesis of a new generation of substituted [4.6.4.6]fenestradienes. Angew. Chem. Int. Ed. 2012, 51, 4379–4382. [Google Scholar] [CrossRef] [PubMed]
- Salacz, L.; Girard, N.; Blond, G.; Suffert, J. Synthesis of Polyheterocyclic Tropones by [2 + 2 + 2 + 1] Carbonylative Cycloaddition of Triynes. Org. Lett. 2018, 20, 3915–3918. [Google Scholar] [CrossRef] [PubMed]
- Pauson, P.L. Tropones and tropolones. Chem. Rev. 1955, 55, 9–136. [Google Scholar] [CrossRef]
- Mori, A.; Kato, N.; Takeshita, H. Preparation and Thermal Properties of New Liquid Crystals with a 2-Benzoyloxy-2,4,6-cycloheptatrien-1-one Core. J. Mater. Chem. 1991, 1, 799–803. [Google Scholar] [CrossRef]
- Takagi, K.; Nishikawa, Y.; Nishioka, N.; Kunisada, H.; Yuki, Y. π-conjugated oligomers containing tropone in the main chain: Synthesis, characterization, and optical properties. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 3927–3937. [Google Scholar] [CrossRef]
- Sugiyasu, K.; Song, C.; Swager, T.M. Aromaticity in tropone-containing polythiophene. Macromolecules 2006, 39, 5598–5600. [Google Scholar] [CrossRef]
- Bentley, R. A fresh look at natural tropolonoids. Nat. Prod. Rep. 2008, 25, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Pietra, F. Revival of Troponoid Chemistry. Acc. Chem. Res. 1979, 12, 132–138. [Google Scholar] [CrossRef]
- Nair, V.; Abhilash, K.G. [8+2] Cycloaddition reactions in organic synthesis. Synlett 2008, 301–312. [Google Scholar] [CrossRef]
- Krafft, M.E.; Cran, J.W. A convenient protocol for the iodination of unsaturated carbonyl compounds with I2 in an aqueous medium. Synlett 2005, 1263–1266. [Google Scholar] [CrossRef]
- Odell, L.R.; Russo, F.; Larhed, M. Molybdenum hexacarbonyl mediated CO gas-free carbonylative reactions. Synlett 2012, 23, 685–698. [Google Scholar] [CrossRef]
- Brummond, K.M.; Chen, D. Mo(CO)6- and [Rh(CO)2Cl]2-Catalyzed Allenic Cyclocarbonylation Reactions of Alkynones: Efficient Access to Bicyclic Dienediones. Org. Lett. 2008, 10, 1154–1156. [Google Scholar] [CrossRef]
- Jeong, N.; Lee, S.J.; Lee, B.Y.; Chung, Y.K. Molybdenium mediated preparation of cyclopentenones. Tetrahedron Lett. 1993, 34, 4027–4030. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Z. Rhodium-Catalyzed [5+2+1] Cycloaddition of Ene− Vinylcyclopropanes and CO: Reaction Design, Development, Application in Natural Product Synthesis, and Inspiration for Developing New Reactions for Synthesis of Eight-Membered Carbocycles. Acc. Chem. Res. 2015, 48, 2288–2296. [Google Scholar] [CrossRef]
- Teng, Y.H.G. Synthesis of Novel Fused Tropones and Colchicinoids Through Rh(I)-Catalyzed [2+2+2+1] Cycloaddition of Triynes with Carbon Monoxide. Ph.D. Thesis, Stony Brook University, Stony Brook, NY, USA, 2011. [Google Scholar]
- Montero-campillo, M.M.; Rodrigez-Otero, J.; Cabaleiro-Lago, E. Theoretical Study of the [2+2+2+1] Cycloaddition Mechanism of Enediynes and Carbon Monoxide Catalyzed by Rhodium. J. Phys. Chem. 2008, 112, 2423–2427. [Google Scholar] [CrossRef]
- Wang, H.; Sawyer, J.R.; Evans, P.A.; Baik, M.H. Mechanistic insight into the diastereoselective rhodium-catalyzed Pauson-Khand reaction: Role of coordination number in stereocontrol. Angew. Chem. Int. Ed. 2008, 47, 342–345. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–d, 2a, 2c–d, 3a, 3c–d and 12 are available from the authors. |




{kind=link}
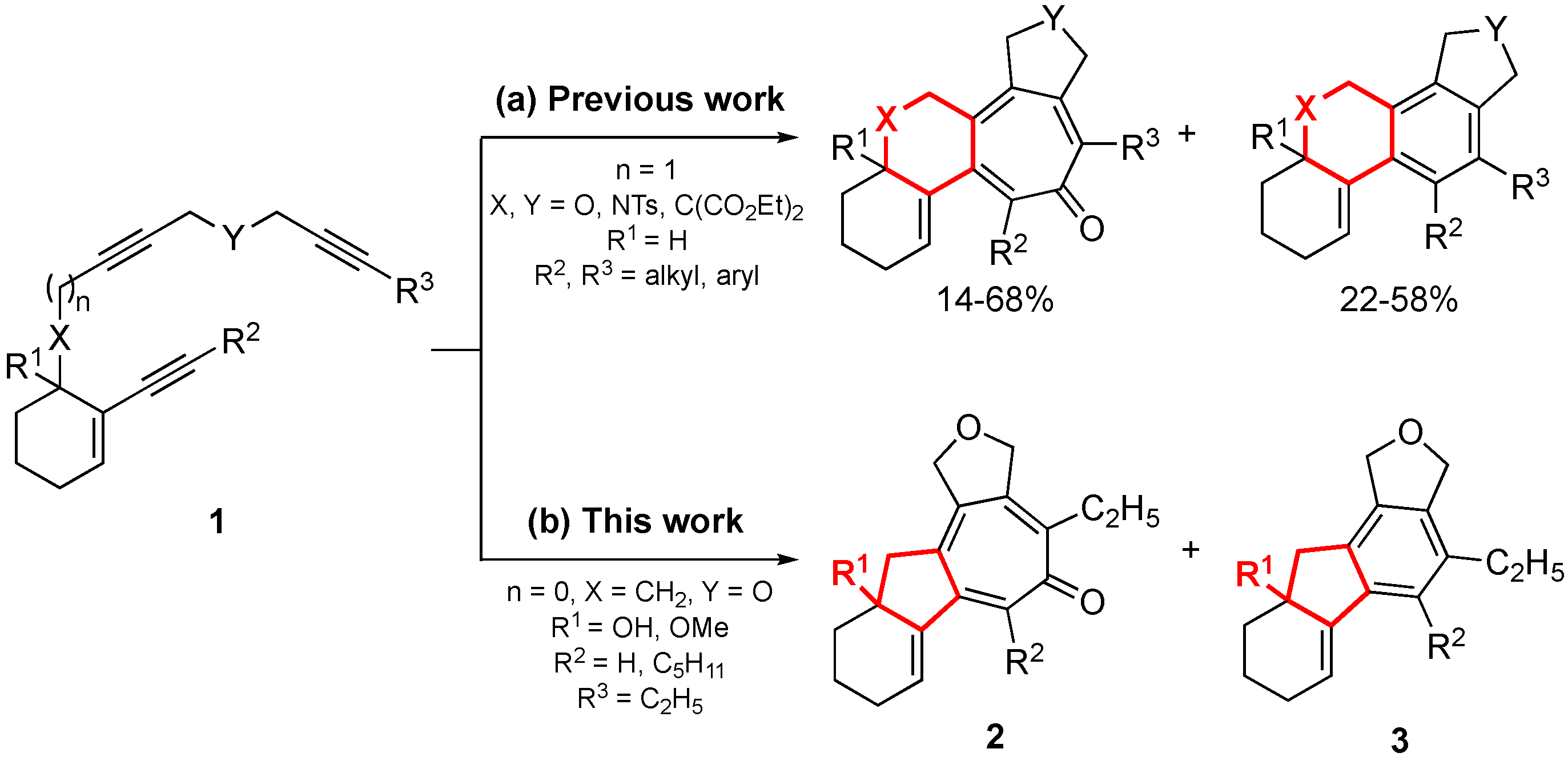{kind=link}
{kind=link}
{kind=link}

| Entry | [Rh] (mol%) | Mo(CO)6 (eq) | Temperature (°C) | Time (h) | Conv. (%) | 2a a | 3a a | 12 a |
|---|---|---|---|---|---|---|---|---|
| 1 b | 5 | 10 | rt | 18 | 100 | 24 (19) | 71 (41) | 5 (5) |
| 2 | 5 | - | rt | 18 | 100 | 25 | 75 | 0 |
| 3 | 25 | - | rt | 0.17 | 100 | (27) | (43) | (30) |
| 4 b | - | 10 | rt | 18 | 90 | 0 | 0 | 90 |
| 5 c | 5 | 10 | 50 | 0.75 | 100 | 25 | 62 | 13 |
| 6 c | 5 | 10 | 100 | 0.5 | 89 | 23 | 54 | 17 |

| Entry | R2 | PCO (atm) | Time (h) | Conversion (%) | 2 a | 3 a | 12, 13 a |
|---|---|---|---|---|---|---|---|
| 1 | 1c | - | 18 | 100 | 2c, 22 | 3c, 73 | 12, 0 |
| 2 | 1d | - | 18 | 100 | 2d, 25 | 3d, 75 | 13, <5 |
| 3 | 1c | 2 | 18 | 100 | 2c, 33 | 3c, 46 | 12, 21 |
| 4 | 1d | 2 | 18 | 100 | 2d, 29 (29) | 3d, 43 (43) | 13, 17 |
| 5 | 1c | 10 | 18 | 100 | 2c, 25 | 3c, 31 | 12, 44 |
| 6 b | 1c | 2 | 18 | 73 | 2c, 26 | 3c, 25 | 12, 22 |
| 7 b | 1c | 2 | 26 | 100 | 2c, 34 (17) | 3c, 32 (24) | 12, 33 (20) |
| 8 c | 1c | 2 | 26 | 90 | 2c, 33 | 3c, 33 | 12, 23 |
| 9 d | 1c | 2 | 26 | 91 | 2c, 36 | 3c, 32 | 12, 23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salacz, L.; Girard, N.; Suffert, J.; Blond, G. Synthesis of Aromatic Rings Embedded in Polycyclic Scaffolds by Triyne Cycloaddition: Competition between Carbonylative and Non-Carbonylative Pathways. Molecules 2019, 24, 595. https://doi.org/10.3390/molecules24030595
Salacz L, Girard N, Suffert J, Blond G. Synthesis of Aromatic Rings Embedded in Polycyclic Scaffolds by Triyne Cycloaddition: Competition between Carbonylative and Non-Carbonylative Pathways. Molecules. 2019; 24(3):595. https://doi.org/10.3390/molecules24030595
Chicago/Turabian StyleSalacz, Laura, Nicolas Girard, Jean Suffert, and Gaëlle Blond. 2019. "Synthesis of Aromatic Rings Embedded in Polycyclic Scaffolds by Triyne Cycloaddition: Competition between Carbonylative and Non-Carbonylative Pathways" Molecules 24, no. 3: 595. https://doi.org/10.3390/molecules24030595
APA StyleSalacz, L., Girard, N., Suffert, J., & Blond, G. (2019). Synthesis of Aromatic Rings Embedded in Polycyclic Scaffolds by Triyne Cycloaddition: Competition between Carbonylative and Non-Carbonylative Pathways. Molecules, 24(3), 595. https://doi.org/10.3390/molecules24030595