2.1. Three-Nucleotide Bulge-Forming RNA Targets
Throughout our research into sequence-specific artificial ribonucleases, the most significant improvements in cleavage rates have consistently resulted from variations of the RNA sequence in the unpaired region of the RNA/PNAzyme complex [
7,
8]. The three-dimensional helical structure of RNA is fascinating, as it is governed by a range of interactions, including canonical and non-canonical base-pairing via hydrogen bonding, as well as nucleobase stacking interactions, which reduce the exposure of hydrophobic surfaces to the polar environment. Structural variation within the confines of helical structures is relatively limited, but stretches of unpaired nucleotides form bulges which add to the diversity of RNA structures as architectural or recognition motifs [
15]. RNA bulges are known to adopt diverse conformations, resulting from the competing interactions between the paired and unpaired nucleobases. Substantial stabilisation of RNA bulges can arise from metal ion interactions, where suitable metal binding pockets are created by backbone distortions that have forced negatively charged phosphate groups in the bulge in close proximity [
15]. For example, due to the strong interactions between Mg
2+ ions and adenine-rich bulges found in group I introns, the RNA has been described as folding around a “metal ion core” [
16].
This study therefore focused on gaining further insights into how changes in the RNA bulge affect their cleavage by PNAzymes. Subjects of this study included RNA targets
1–
15, which contain two recognition motifs for PNAzyme
I that are identical to those investigated in our previous studies [
12] comprising seven canonical base pairs on one side and a wobble base pair closing the bulge, followed by three Watson-Crick pairs on the other side (
Table 1). We have previously reported substantial differences between RNAs forming 3- and 4-nucleotide bulges when bound to and cleaved by Zn
2+-dependent PNAzymes [
14], but little is known about 3-nucleotide RNA bulges and Cu
2+ PNAzymes, which are the most efficient and most selective artificial nucleases reported.
Overall, the rate of the PNAzyme-promoted cleavage of 3-nucleotide RNA bulges in the presence of Cu
2+ ions was substantially lower than the rates observed with 4-nucleotide RNA bulges. The fastest 4-nucleotide bulge sequences have previously been shown to give half-lives of around 20–30 min [
12,
13], while the 3-nucleotide bulge-forming RNA sequences were cleaved with approximately 14–24-h half-lives at best. Nonetheless, the cleavage of RNA was highly site-selective and displayed similar tendencies to the previously studied 4-nucleotide bulge systems regarding the nucleotide preferences at specific positions [
12].
Interestingly, while the general bulge sequence APyPuA (Py referring to pyrimidine and Pu to purine bases) displayed the highest rates of cleavage for 4-nucleotide bulges [
12], a similar correlation was observed with the 3-nucleotide bulge systems. Unequivocally, adenosine is the nucleotide best accepted next to the wobble base pair in the 3-nucleotide bulge systems (RNA
1–
4,
6,
10–
12;
Table 1). The presence of any other nucleotide at that position (RNA
5,
7–
9,
13–
15;
Table 1) resulted in the extent of RNA cleavage after 24 h being barely above the detection limit. Furthermore, additional similarities can be noted, as either of the purine bases, adenine or guanine (RNA
1,
3,
6;
Table 1), were preferred over pyrimidine bases (RNA
2,
4,
10–
12;
Table 1) at the adjacent position. While previously, APyPuA bulges displayed the highest reaction rates, AAPuA resulted in only a slightly lower cleavage efficiency. Likewise, UAA and APuA bulges were the most readily cleaved in the 3-nucleotide bulge system. The overall sequence trends seen with 4-nucleotide bulges appear to be similar for 3-nucleotide bulges. The cleavage of 3-nucleotide RNA bulges occurred in a highly site-selective manner, producing a 2′,3′-cyclic phosphate as the longer fragment, as identified by mass spectrometry. Most importantly, the purine-adenosine cleavage site was preserved and seems to be a minimum requirement for RNA cleavage. Nevertheless, the dramatic decrease in the reaction rate highlights the structural importance of the additional adenosine in 4-nucleotide bulges.
In general, the preservation of continuous stacking in the double helix is a powerful driving force known to define the structure and interactions of RNA bulges [
15]. Moreover, molecular dynamics simulations of an RNA that forms an AAAA bulge closed with a wobble base pair have suggested that the first bulge adenosine on the 5’ side of the bulge is involved in cross-strand stacking interactions, introducing significant rigidity to the structure, whereas the wobble on the other side of the bulge adds flexibility [
17]. Therefore, the impact that the bulge adenosine stacking interactions have on the overall conformation and rigidity of the bulge could be substantial.
2.2. Modified Four-Nucleotide Bulge-Forming RNA Targets
In order to gain additional insight into the mechanism and sequence preferences of the efficient cleavage of 4-nucleotide bulge-forming RNA targets, we then performed studies where the cleavage site of the RNA forming an AUAA bulge was modified. The natural, unmodified AUAA bulge (RNA
16,
Table 2) is cleaved to a substantial extent in the presence of PNAzyme
I and Cu
2+ ions at pH 7 after only a 1-h reaction time. However, if the adenosine providing the 2′-hydroxyl nucleophile is replaced with an unsubstituted purine riboside lacking the exocyclic amine (RNA
17,
Table 2) to give an AUPurA bulge, the reaction rate drops substantially, increasing the half-life from about 30 min to around 24 h. These findings suggest that the amino group may coordinate to the Cu
2+ ion and thereby have a role in the positioning of the catalytic ion in critical proximity to the cleavage site. From our previous studies, it is clear that the adenine base can be replaced with guanine without a significant loss in activity, suggesting that the carbonyl functionality on the guanosine can play a similar role to the exocyclic amine on the adenosine. It is also plausible that the pKa of the hydrated metal ion is affected. This would be especially crucial if the mechanism of cleavage involves protonation of the 5′-hydroxyl leaving group by a water molecule bound to the copper ion, as has been suggested for the cleavage of metal aquo ions [
18].
Moreover, ribose modifications were introduced by replacing the central adenosine with a deoxyadenosine nucleotide (RNA
18,
Table 2) or with a 2′-alkyl modified adenosine (RNA
19,
Table 2). As the 2′-hydroxyl group supplies the nucleophile for the transesterification reaction, these substitutions were expected to result in diminished catalytic activity, which was demonstrated. This is also consistent with the previously reported observation that the immediate product formed upon cleavage by PNAzyme
I is the 2′,3′-cyclic phosphate [
12]. In addition, these results further substantiate the almost complete selectivity of cleavage at a single site, since not even if this site is blocked is the RNA cleaved at an alternate site. Moreover, the possibility to form an RNA/PNAzyme complex with a blocked cleavage site may also allow for the determination of the crystal structure of the complex with Cu
2+ bound to the neocuproine.
It has been suggested that the activation of phosphodiesters in an RNA strand can be achieved by structural alteration obtained when nucleobases interact with polyaromatic molecules. An example is the introduction of acridine groups in the recognising strand, which makes the RNA target more susceptible to cleavage by lanthanide ions in the vicinity of the acridine moieties and hence gives a higher selectivity. This “pin-point” activation further substantiates that not only the cleaver, but also the structure of the RNA substrate, are crucial elements of the cleavage reaction [
19,
20]. In order to evaluate if such an effect was partially responsible for the selectivity and high cleavage rate obtained with PNAzyme
I, we investigated whether the corresponding phenanthrene conjugate would give substantial cleavage of RNA in the presence of external Cu
2+ ions. Although it cannot be completely excluded, the absence of cleavage with the PNA-phenanthrene conjugate (
Table 2, RNA
16 (
Y), last entry) suggests that pin-point activation does not play a major role in the case of these Cu
2+ PNAzymes.
2.3. PNAzyme II Containing an Extended Recognition Arm
We had asked ourselves if the GCCC hybridising stem, which is the shorter recognition arm of the RNA/PNAzyme complex, retains the duplex conformation during the cleavage of the RNA target. In order to understand the role of this short recognition arm, a new artificial ribonuclease was designed by introducing an extension of two adenine-PNA units to the amino terminus of PNAzyme I to give PNAzyme II. The principal aim of this investigation was to see whether the PNAzyme activity is retained upon elongation of the RNA/PNAzyme complex. A result to the contrary would suggest that the shorter hybridised region potentially undergoes a structural change critical for the catalytic activity, which would be inhibited by stabilisation of the double strand due to additional base-pairing.
Initially, the ability of the two PNAzymes to promote the cleavage of 4-nucleotide bulge-forming RNAs
16 and
20–
22 was compared. PNAzyme
I-promoted cleavage of RNA bulge sequences AUAA and ACAA (RNA
16 and
22) occurs with half-lives below 30 min in the presence of Cu
2+ ions at pH 7, while an AAAA bulge (RNA
21) is cleaved at a slightly lower rate and an AAUA bulge is essentially non-cleavable (RNA
20,
Table 3). Upon complexation of these RNAs with PNAzyme
II, the extension introduced to the new PNAzyme presents a dangling overhang in relatively close proximity to the cleavage site. The overall trend of cleavage rate dependence on the bulge sequence remained similar to that seen with PNAzyme
I. Although still displaying efficient cleavage at the same site, the overhang in PNAzyme
II was shown to reduce the cleavage rates, as illustrated by the decrease in the extent of RNA cleavage after 30 min (
Table 3). These observations further demonstrate the specificity of this system, as the delicate molecular environment around the cleavage site can be disturbed by overhanging nucleotides in the case of off-target RNA sequences with incomplete recognition arms. This may become important as it could further increase the specificity of PNAzymes.
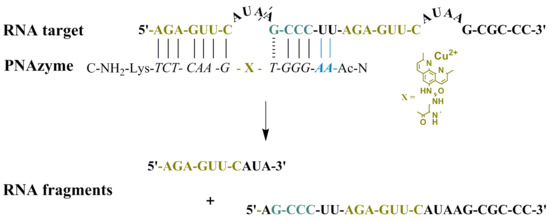
Moreover, the cleavage of an extended RNA target was studied (RNA
23,
Figure 1), where the extension present in PNAzyme
II is able to form additional Watson-Crick base pairs, with the UU sequence in the RNA preceding the short recognition arm. As such, compared to the complex with RNA
16, four additional hydrogen bonds will be formed with RNA
23, stabilising the duplex stem (
Figure 1, Scenario 1). In addition to containing the RNA
16 sequence with an extended short recognition arm, RNA
23 also contains a competing long recognition arm, the potential to form the fastest cleaved bulge and a mismatch-containing sequence that otherwise resembles the short recognition arm (
Figure 1, Scenario 2).
As previously shown, a single mismatch in the short arm of RNA
16 leads to a very low cleavage rate with PNAzyme
I, [
12] and the cleavage of RNA
23 occurs exclusively at the bulge formed between the matching recognition arms, producing fragments
1A and
1B (
Figure 1). This demonstrates excellent mismatch discrimination. While PNAzyme
I-promoted cleavage of this long RNA
23 target is qualitatively analogous to the cleavage of RNA
16, the cleavage of RNA
23 seemed to occur at a somewhat lower rate (
Table 4). In contrast, the extended PNAzyme
II-promoted cleavage of RNA
23 occurred at a higher rate than with PNAzyme
I. In fact, the improved rate was essentially identical to that of PNAzyme
I-promoted cleavage of RNA
16. Therefore, not only did the extended recognition arm leave the catalytic activity undisturbed, but it allowed for the fastest cleavage rate to be retained for the first time on a longer, more complex RNA target which resembles a more likely natural target.
Moreover, the cleavage of another long RNA sequence (RNA
24,
Figure 2) was studied, where the same short recognition arm was only extended by one nucleobase instead of two. In addition, RNA
24 contained an alternative set of recognition arms further along the sequence, surrounding a bulge sequence that renders the system catalytically inactive [
12]. Crucially, now, the alternative binding scenario provides more competition than it did in the case of RNA
23, due to the absence of the mismatch, while a competing cleavage site is missing and thus only site-specific cleavage producing fragments
1A and
1B can occur. As expected, the extent of PNAzyme
II promoted RNA cleavage after 30 min decreased (RNA
24,
Table 4), possibly due to competitive binding to the non-productive site and/or due to lower activity with PNA overhang.
Finally, the ability to give turnover of the substrate (and thus exhibit true catalytic behaviour) is a crucial feature of an artificial enzyme. As such, the overall potency of an artificial ribonuclease depends upon its overall affinity to the target, but also on its ability to release the cleaved RNA fragments, which then allows for it to bind to and trigger the cleavage of the next target. Thus, the potency of a catalytic antisense oligonucleotide could in fact be reduced by overly strong binding to the target. As efficient turnover of RNA
16 by PNAzyme
I has been previously demonstrated [
12], it then became critical to investigate whether the additional base-pairs in the RNA
23/PNAzyme
II complex may enhance the binding to such a degree as to impair turnover. This was shown not to be the case in experiments with sub-stoichiometric PNAzyme
II. Multiple turnover was clearly demonstrated after a 24-h period with a 10-fold excess of RNA
23 (
Figure 3).







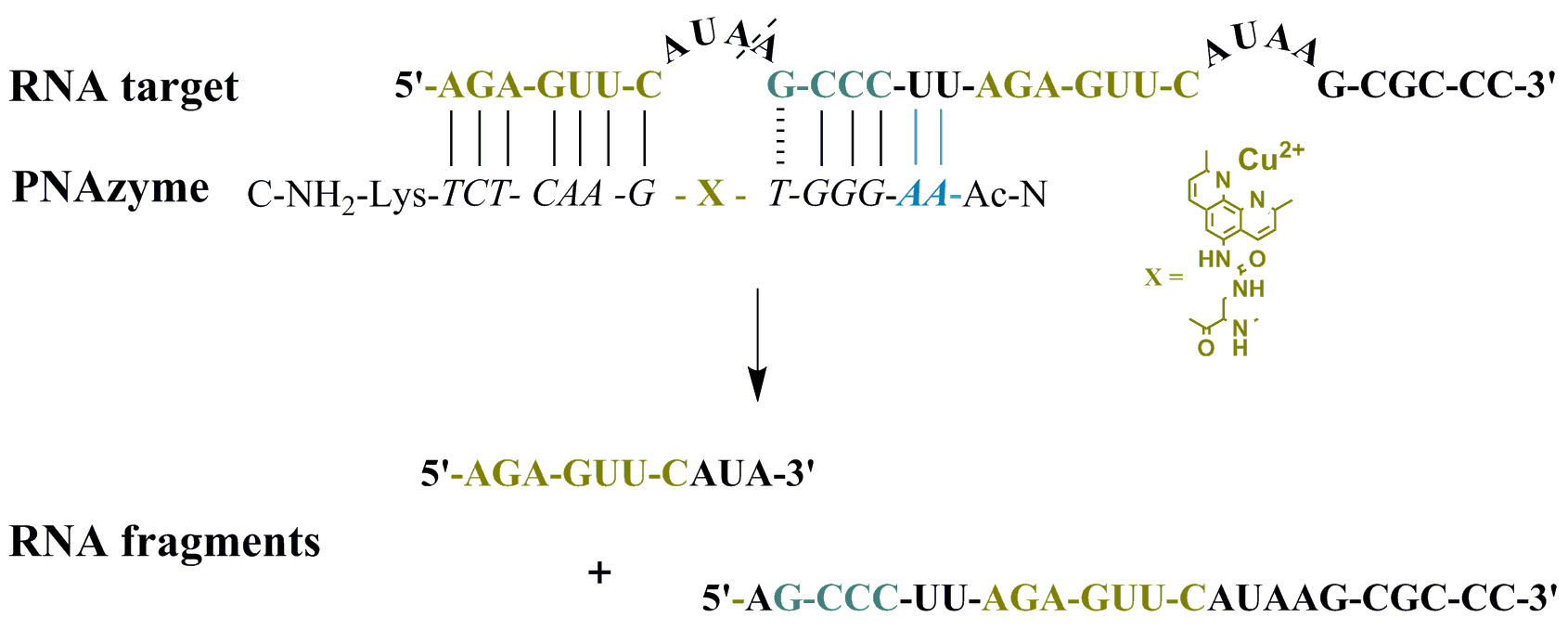{kind=link}
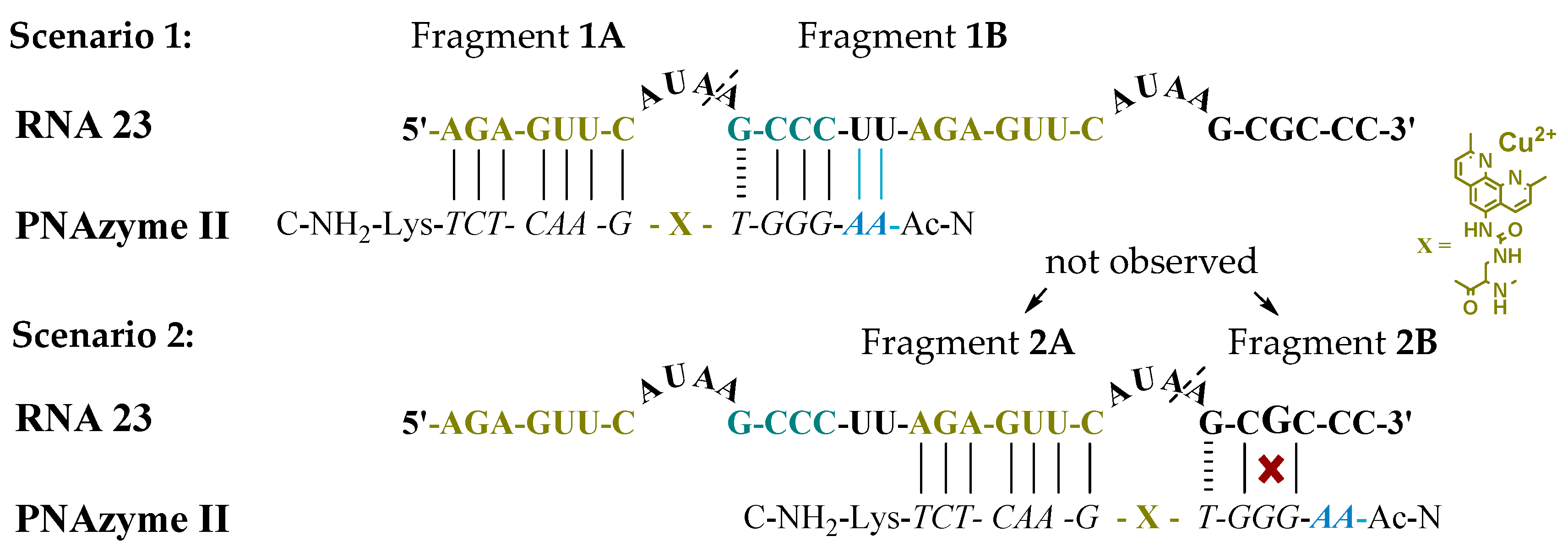{kind=link}
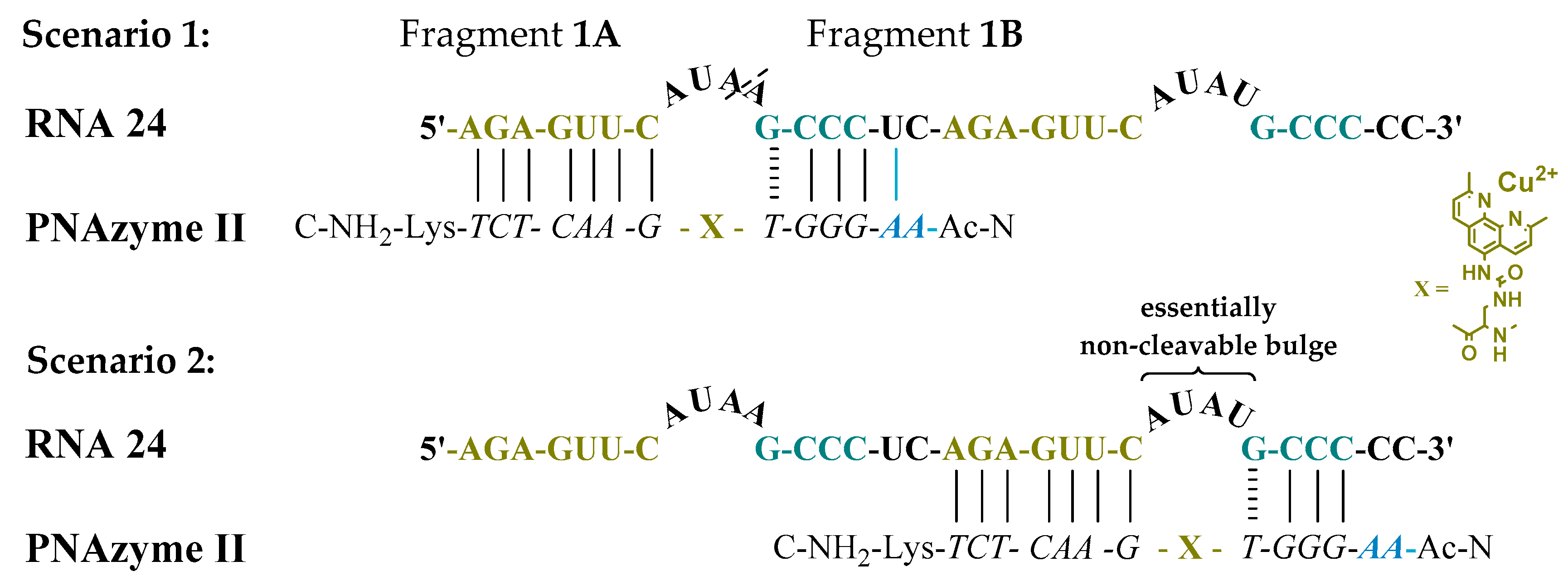{kind=link}
{kind=link}


