Application of a Pillared-Layer Zn-Triazolate Metal-Organic Framework in the Dispersive Miniaturized Solid-Phase Extraction of Personal Care Products from Wastewater Samples

 , ,
, ,  , ,
, , 
Abstract
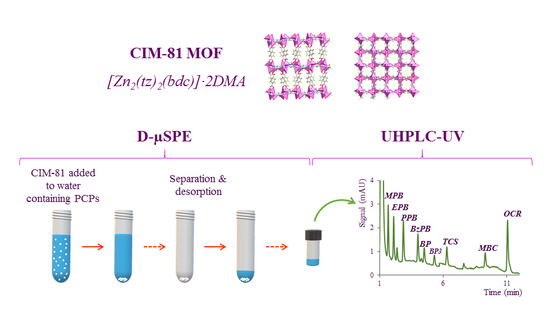
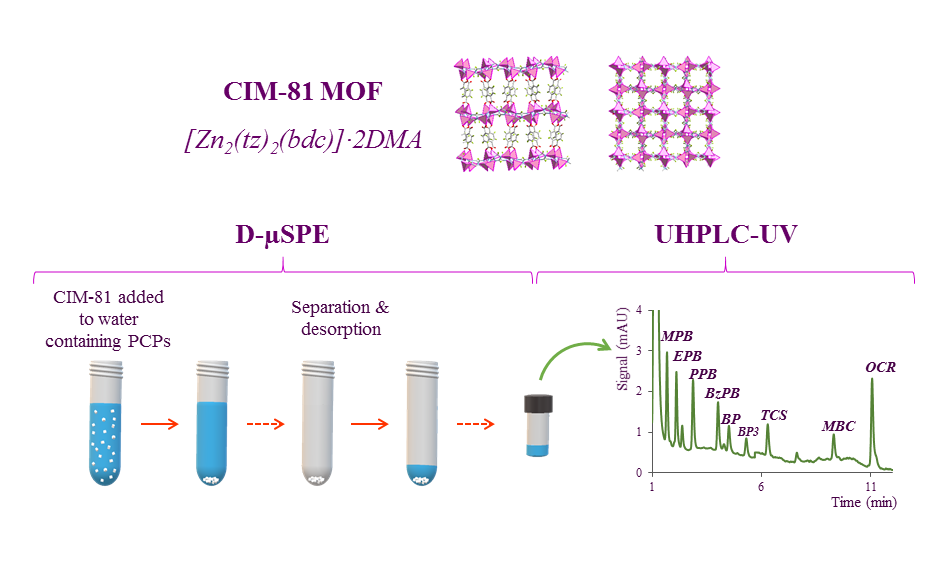
:
1. Introduction
2. Results and Discussion
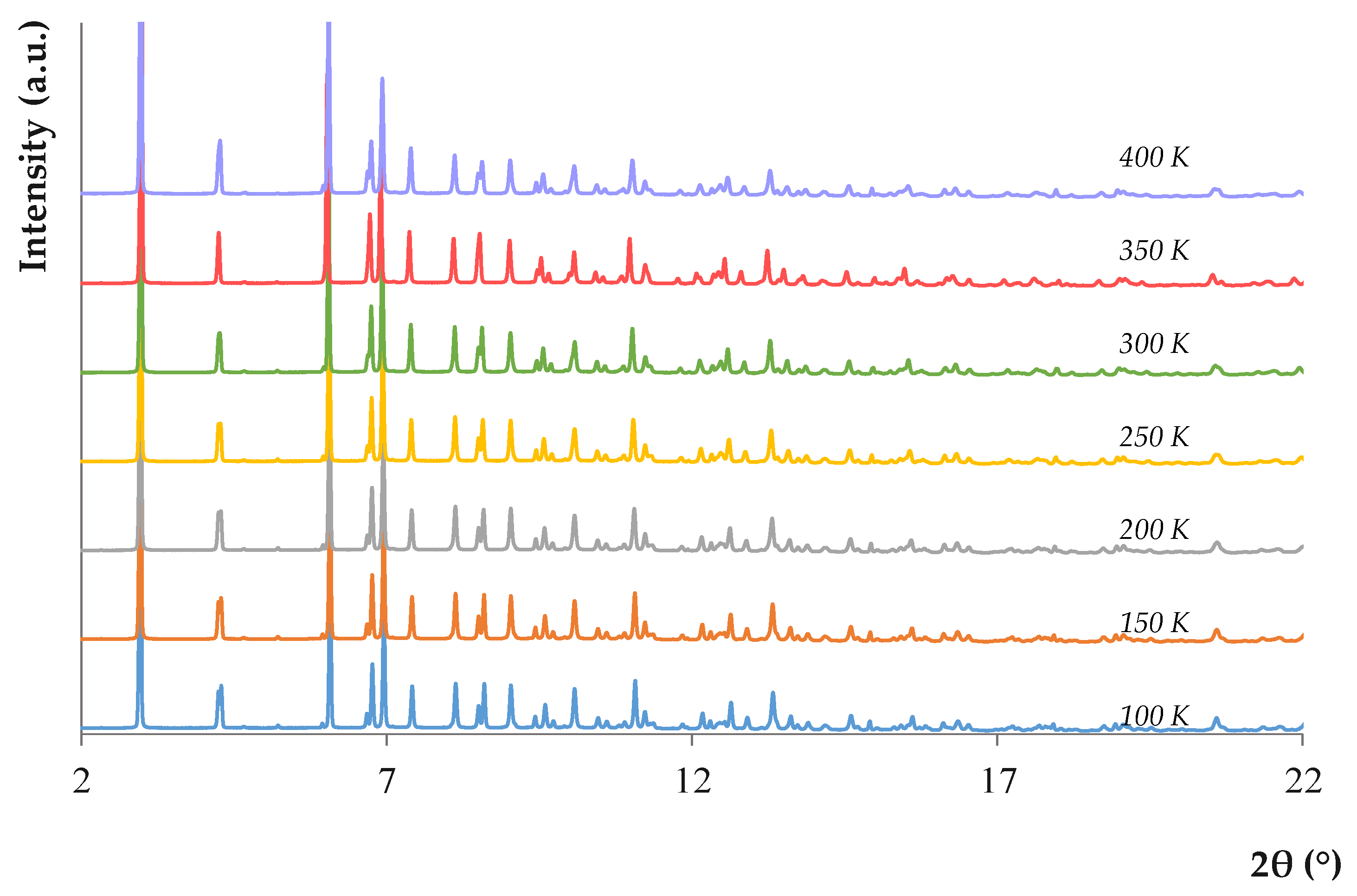
2.1. Characterization of the CIM-81 MOF
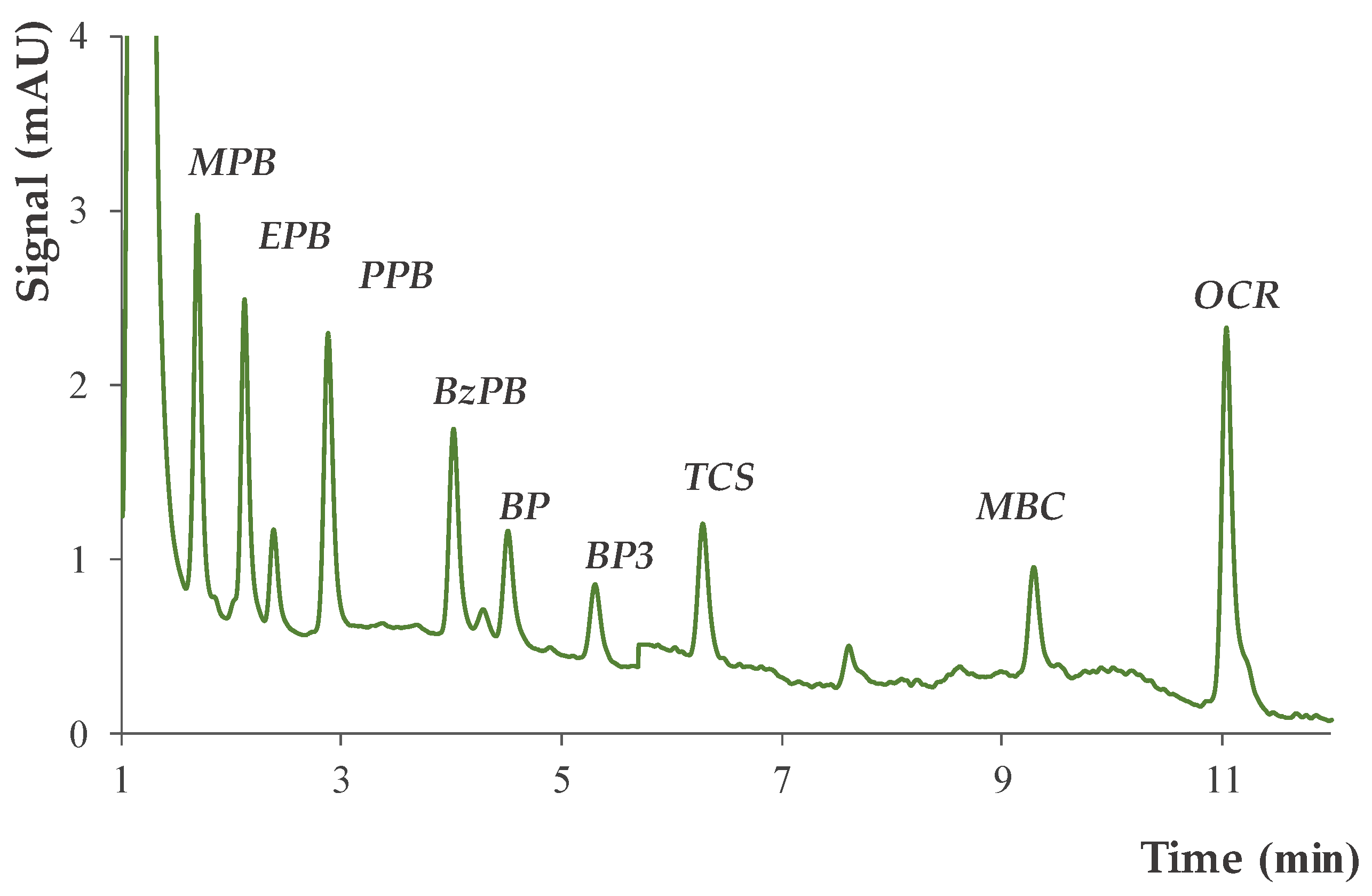
2.2. Chromatographic Method
2.3. Optimization of the D-µSPE Procedure
2.3.1. Preliminary Studies in the D-µSPE-UHPLC-UV Method
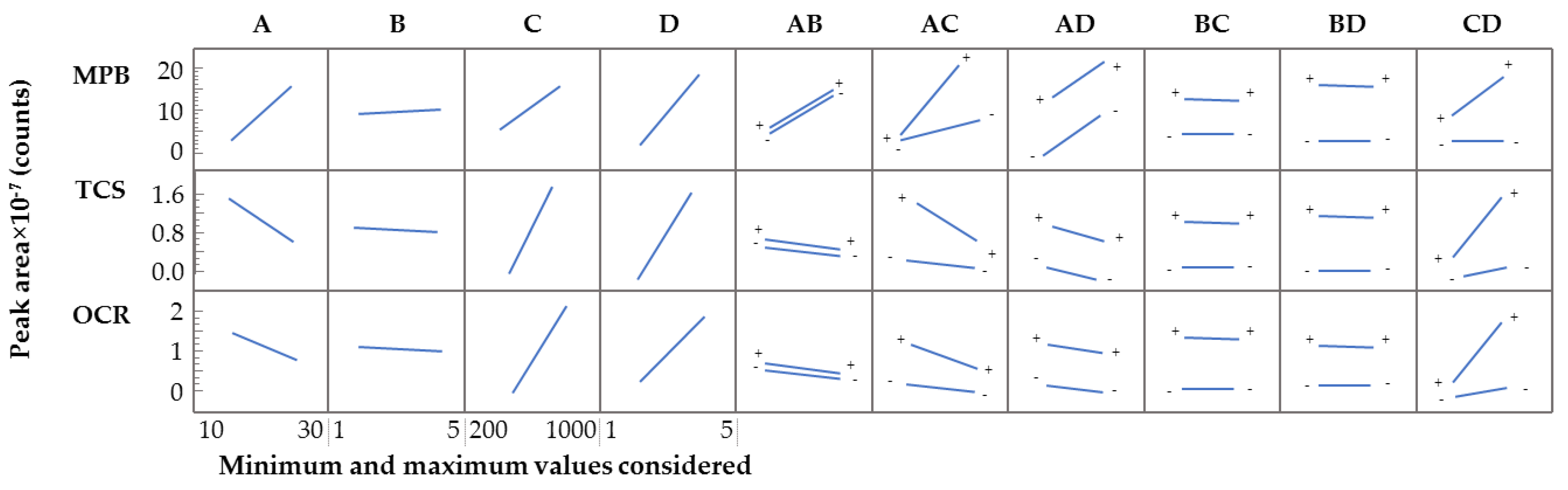
2.3.2. Screening Optimization Study for the D-µSPE-UHPLC-UV Method
2.3.3. Doehlert Design for the D-µSPE-UHPLC-UV Method
2.4. Analytical Performance of the Optimized D-µSPE-UHPLC-UV Method
2.5. Reuse of the CIM-81 MOF in the D-µSPE-UHPLC-UV Method
2.6. Analysis of Samples
3. Experimental Section
3.1. Standards, Reagents and Materials
3.2. Sample Collection
3.3. Instrumentation
3.4. Procedures
3.4.1. Synthesis of MOF
3.4.2. Chromatographic Method
3.4.3. Dispersive Miniaturized Solid-Phase Extraction (D-µSPE) Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| BET | Brunauer, Emmet and Teller |
| BP | benzophenone |
| BP3 | benzophenone-3 |
| BzPB | benzylparaben |
| DMA | N-N˗dimethylacetamide |
| D-µSPE | dispersive miniaturized solid phase extraction |
| EPB | ethylparaben |
| GAC | Green Analytical Chemistry |
| LLE | liquid-liquid extraction |
| MBC | 3-(4-methylbenzylidene) camphor |
| MOF | metal-organic frameworks |
| MPB | methylparaben |
| OCR | octocrylene |
| PPB | propylparaben |
| PCPs | personal care products |
| SBSE | stir-bar sorptive microextraction |
| SCSE | stir-cake sorptive microextraction |
| SPE | solid-phase extraction |
| SPME | solid-phase microextraction |
| TCS | triclosan |
| µSPE | miniaturized solid phase extraction |
| UHPLC | ultra-high performance liquid chromatography |
References
- Giulivo, M.; López de Alda, M.; Capri, E.; Barceló, D. Human exposure to endocrine disrupting compounds: Their role in reproductive systems, metabolic syndrome and breast cancer. A review. Environ. Res. 2016, 151, 251–264. [Google Scholar] [CrossRef]
- Lapworth, D.J.; Baran, N.; Stuart, M.E.; Ward, R.S. Emerging organic contaminants in groundwater: A review of sources, fate and occurrence. Environ. Poll. 2012, 163, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Armenta, S.; Garrigues, S.; de la Guardia, M. The role of green extraction techniques in Green Analytical Chemistry. TrAC-Trends Anal. Chem. 2015, 71, 2–8. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J.; Szczepańska, N.; de la Guardia, M.; Namieśnik, J. Miniaturized solid-phase extraction techniques. TrAC-Trends Anal. Chem. 2015, 73, 19–38. [Google Scholar] [CrossRef]
- Carasek, E.; Morés, L.; Merib, J. Basic principles, recent trends and future directions of microextraction techniques for the analysis of aqueous environmental samples. Trends Environ. Anal. Chem. 2018, 19, e0060. [Google Scholar] [CrossRef]
- Pacheco-Fernández, I.; Gutiérrez-Serpa, A.; Rocío-Bautista, P.; Pino, V. Molecularly imprinted polymers as promising sorbents in SPME applications. In Solid-Phase Microextraction: Advances in Research and Applications, 1st ed.; Verreau, W., Baril, G., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2017; pp. 147–168. [Google Scholar]
- Wen, Y.; Chen, L.; Li, J.; Liu, D.; Chen, L. Recent advances in solid-phase sorbents for sample preparation prior to chromatographic analysis. TrAC-Trends Anal. Chem. 2014, 59, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Lucena, R.; Simonet, B.M.; Cárdenas, S.; Valcárcel, M. Potential of nanoparticles in sample preparation. J. Chromatogr. A 2011, 1218, 620–637. [Google Scholar] [CrossRef]
- Wang, Y.; Rui, M.; Lu, G. Recent applications of metal-organic frameworks in sample pretreatment. J. Sep. Sci. 2018, 41, 180–194. [Google Scholar] [CrossRef]
- Yaghi, O.M.; Li, H. Hydrothermal synthesis of a metal-organic framework containing large rectangular channels. J. Am. Chem. Soc. 1995, 117, 10401–10402. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; Pacheco-Fernández, I.; Pasán, J.; Pino, V. Are metal-organic frameworks able to provide a new generation of solid-phase microextraction coatings?–A review. Anal. Chim. Acta 2016, 939, 26–41. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; González-Hernández, P.; Pino, V.; Pasán, J.; Afonso, A.M. Metal-organic frameworks as novel sorbents in dispersive-based microextraction approaches. TrAC-Trends Anal. Chem. 2017, 90, 114–134. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z. Metal-organic frameworks as stationary phase for application in chromatographic separation. J. Chromatogr. A 2017, 1530, 1–18. [Google Scholar] [CrossRef]
- Pacheco-Fernández, I.; González-Hernández, P.; Pasán, J.; Ayala, J.H.; Pino, V. The rise of metal-organic frameworks in analytical chemistry. In Handbook of Smart Materials in Analytical Chemistry, 1st ed.; de la Guardia, M., Esteve-Turrillas, F.A., Eds.; Wiley: Weinheim, Germany, 2019; Volume 1, pp. 463–502. [Google Scholar]
- González-Hernández, P.; Gutiérrez-Serpa, A.; Rocío-Bautista, P.; Pasán, J.; Ayala, J.H.; Pino, V. Micro-solid-phase extraction using metal-organic frameworks. In Metal Organic Frameworks: Properties and Applications, 1st ed.; Mittal, V., Ed.; Central West Publishing: Orange, Australia, 2019; in press. [Google Scholar]
- Li, X.; Xing, J.; Chang, C.; Wang, X.; Bai, Y.; Yan, X.; Liu, H. Solid-phase extraction with the metal–organic framework MIL-101(Cr) combined with direct analysis in real time mass spectrometry for the fast analysis of triazine herbicides. J. Sep. Sci. 2014, 37, 1489–1495. [Google Scholar] [CrossRef]
- Lu, N.; Wang, T.; Zhao, P.; Zhang, L.; Lun, X.; Zhang, X.; Hou, X. Experimental and molecular docking investigation on metal-organic framework MIL-101(Cr) as a sorbent for vortex assisted dispersive micro-solid-phase extraction of trace 5-nitroimidazole residues in environmental water samples prior to UPLC-MS/MS analysis. Anal. Bioanal. Chem. 2016, 408, 8515–8528. [Google Scholar] [CrossRef]
- Taima-Mancera, I.; Rocío-Bautista, P.; Pasán, J.; Ayala, J.H.; Ruiz-Pérez, C.; Afonso, A.M.; Lago, A.B.; Pino, V. Influence of Ligand Functionalization of UiO-66-Based Metal-Organic Frameworks When Used as Sorbents in Dispersive Solid-Phase Analytical Microextraction for Different Aqueous Organic Pollutants. Molecules 2018, 23, 2869. [Google Scholar] [CrossRef]
- Cao, X.; Jiang, Z.; Wang, S.; Hong, S.; Li, H.; Zhang, C.; Shao, Y.; She, Y.; Jin, F.; Jin, M.; et al. Metal-organic framework UiO-66 for rapid dispersive solid phase extraction of neonicotinoid insecticides in water samples. J. Chromatogr. B 2017, 1077–1078, 92–97. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; Pino, V.; Pasán, J.; López-Hernández, I.; Ayala, J.H.; Ruiz-Pérez, C.; Afonso, A.M. Insights in the analytical performance of neat metal-organic frameworks in the determination of pollutants of different nature from waters using dispersive miniaturized solid-phase extraction and liquid chromatography. Talanta 2018, 179, 775–783. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; Pino, V.; Ayala, J.H.; Pasán, J.; Ruiz-Pérez, C.; Afonso, A.M. The metal-organic framework HKUST-1 as efficient sorbent in a vortex-assisted dispersive micro solid-phase extraction of parabens from environmental waters, cosmetic creams and human urine. Talanta 2015, 139, 13–20. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; Pino, V.; Ayala, J.H.; Pasán, J.; Ruiz-Pérez, C.; Afonso, A.M. A magnetic-based dispersive micro-solid-phase extraction method using the metal-organic framework HKUST-1 and ultra-high-performance liquid chromatography with fluorescence detection for determining polycyclic aromatic hydrocarbons in waters and fruit tea infusions. J. Chromatogr. A 2016, 1436, 42–50. [Google Scholar]
- Ge, D.; Lee, H.K. Water stability of zeolite imidazolate framework 8 and application to porous membrane-protected micro-solid-phase extraction of polycyclic aromatic hydrocarbons from environmental water samples. J. Chromatogr. A 2011, 1218, 8490–8495. [Google Scholar] [CrossRef]
- Su, H.; Lin, Y.; Wang, Z.; Wong, Y.-L.E.; Chen, X.; Chan, T.-W.D. Magnetic metal–organic framework–titanium dioxide nanocomposite as adsorbent in the magnetic solid-phase extraction of fungicides from environmental water samples. J. Chromatogr. A 2016, 1466, 21–28. [Google Scholar] [CrossRef]
- Lv, Z.; Sun, Z.; Song, C.; Lu, S.; Chen, G.; You, J. Sensitive and background-free determination of thiols from wastewater samples by MOF-5 extraction coupled with high-performance liquid chromatography with fluorescence detection using a novel fluorescence probe of carbazole-9-ethyl-2-maleimide. Talanta 2016, 161, 228–237. [Google Scholar] [CrossRef]
- Zhou, Q.; Lei, M.; Li, J.; Liu, Y.; Zhao, K.; Zhao, D. Magnetic solid phase extraction of N- and S-containing polycyclic aromatic hydrocarbons at ppb levels by using a zerovalent iron nanoscale material modified with a metal organic framework of type Fe@MOF-5, and their determination by HPLC. Microchim. Acta 2017, 184, 1029–1036. [Google Scholar] [CrossRef]
- González-Sálamo, J.; González-Curbelo, M.A.; Hernández-Borges, J.; Rodríguez-Delgado, M.A. Use of Basolite® F300 metal-organic framework for the dispersive solid-phase extraction of phthalic acid esters from water samples prior to LC-MS determination. Talanta 2019, 195, 236–244. [Google Scholar] [CrossRef]
- Tahmasebi, E.; Masoomi, M.Y.; Yamini, Y.; Morsali, A. Application of a Zn(II) based metal-organic framework as an efficient solid-phase extraction sorbent for preconcentration of plasticizer compounds. RSC Adv. 2016, 6, 40211–40218. [Google Scholar] [CrossRef]
- Safari, M.; Yamini, Y.; Masoomi, M.Y.; Morsali, A.; Mani-Varnosfaderani, A. Magnetic metal-organic frameworks for the extraction of trace amounts of heavy metal ions prior to their determination by ICP-AES. Microchim. Acta 2017, 184, 1555–1564. [Google Scholar] [CrossRef]
- Wang, T.; Wang, J.; Zhang, C.; Yang, Z.; Dai, X.; Cheng, M.; Hou, X. Metal–organic framework MIL-101(Cr) as a sorbent of porous membrane-protected micro-solid phase extraction for the analysis of six phthalate esters from drinking water: A combination of experimental and computational study. Analyst 2015, 140, 5308–5316. [Google Scholar] [CrossRef]
- Rocío-Bautista, P.; Pino, V.; Ayala, J.H.; Ruiz-Pérez, C.; Vallcorba, O.; Afonso, A.M.; Pasán, J. A Green metal-organic framework to monitor water contaminants. RSC Adv. 2018, 8, 31304–31310. [Google Scholar] [CrossRef]
- Kitaura, R.; Iwahori, F.; Matsuda, R.; Kitagawa, S.; Kubota, Y.; Takata, M.; Kobayashi, T.C. Rational Design and Crystal Structure Determination of a 3-D Metal-Organic Jungle-Gym-like Open Framework. Inorg. Chem. 2004, 43, 6522–6524. [Google Scholar] [CrossRef]
- Senthilkumar, S.; Goswami, R.; Obasi, N.L.; Neogi, S. Construction of Pillar-Layer Metal−Organic Frameworks for CO2 Adsorption under Humid Climate: High Selectivity and Sensitive Detection of Picric Acid in Water. ACS Sustain. Chem. Eng. 2017, 5, 11307–11315. [Google Scholar] [CrossRef]
- Du, M.; Li, C.-P.; Liu, C.-S.; Fang, S.-M. Design and construction of coordination polymers with mixed-ligand synthetic strategy. Coord. Chem. Rev. 2013, 257, 1282–1305. [Google Scholar] [CrossRef]
- Zhai, Q.-G.; Bai, N.; Li, S.; Bu, X.; Feng, P. Design of Pore Size and Functionality in Pillar-Layered Zn-Triazolate-Dicarboxylate Frameworks and Their High CO2/CH4 and C2 Hydrocarbons/CH4 Selectivity. Inorg. Chem. 2015, 54, 9862–9868. [Google Scholar] [CrossRef]
- Hu, X.-L.; Gong, Q.-H.; Zhong, R.-L.; Wang, X.-L.; Qin, C.; Wang, H.; Li, J.; Shao, K.-Z.; Su, Z.-M. Evidence of amine-CO2 interactions in two pillared-layer MOFs probed by X-ray crystallography. Chem. Eur. J. 2015, 21, 7238–7244. [Google Scholar] [CrossRef]
- Araujo, P.; Janagap, S. Doehlert uniform shell designs and chromatography. J. Chromatogr. B 2012, 910, 14–21. [Google Scholar] [CrossRef]
- Fauth, F.; Peral, I.; Popescu, C.; Knapp, M. The new Material Science Powder Diffraction beamline at ALBA Synchrotron. Powder Diffr. 2013, 28, S360–S370. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCP | (Slope ± Sba) × 10˗4 | R2 b | Sy/x c × 10−3 | LOD/LOQ d (µg·L−1) | EF e | Conc. Level: 15 µg·L−1 | Conc. Level: 45 µg·L−1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RSDf (%) intra-day/inter-day | RRg (%) | EFh | RSDf (%) intra-day/inter-day | RRg (%) | EFh | ||||||
| MPB | 207 ± 6 | 0.9964 | 5.1 | 0.7/2.3 | 5.3 | 5.1/7.2 | 122 | 9.1 | 2.3/7.1 | 108 | 6.6 |
| EPB | 233 ± 5 | 0.9973 | 5.0 | 1.0/3.3 | 6.8 | 5.0/6.5 | 125 | 7.2 | 2.6/5.1 | 111 | 7.1 |
| PPB | 247 ± 7 | 0.9963 | 6.2 | 0.8/2.7 | 8.1 | 5.5/9.4 | 108 | 8.1 | 5.6/8.5 | 97.7 | 7.7 |
| BzPB | 180 ± 4 | 0.9976 | 3.7 | 0.9/3.0 | 6.7 | 6.6/8.3 | 121 | 7.4 | 3.7/7.0 | 115 | 7.5 |
| BP | 121 ± 4 | 0.9957 | 3.3 | 1.5/5.0 | 3.1 | 8.7/10 | 112 | 3.6 | 4.4/9.4 | 80.9 | 2.6 |
| BP3 | 139 ± 4 | 0.9961 | 3.6 | 1.2/4.0 | 5.4 | 6.2/13 | 118 | 6.6 | 6.7/9.4 | 97.8 | 5.3 |
| TCS | 36 ± 1 | 0.9971 | 0.80 | 1.0/3.3 | 5.7 | 7.2/11 | 94.1 | 12 | 4.1/8.4 | 92.8 | 7.2 |
| MBC | 116 ± 3 | 0.9956 | 3.2 | 1.4/4.7 | 3.3 | 4.6/6.6 | 120 | 3.6 | 2.6/8.2 | 99.5 | 3.2 |
| OCR | 350 ± 4 | 0.9992 | 4.0 | 0.5/1.7 | 29 | 6.6/12 | 112 | 34 | 8.0/6.0 | 96.2 | 29 |
| PCP | Sample 1 | Sample 2 | Sample 3 * | |||
|---|---|---|---|---|---|---|
| Spiked Level: 15 µg·L−1 | Spiked Level: 45 µg·L−1 | |||||
| RRa (%) | EFb | RRa (%) | EFb | |||
| MPB | 7.4 ± 0.8 | 7.6 ± 0.9 | 101 | 8.0 | 113 | 6.9 |
| EPB | 13.2 ± 0.3 | n.d. | 122 | 7.0 | 119 | 7.6 |
| PPB | n.d. | n.d. | 120 | 9.1 | 112 | 8.9 |
| BzPB | n.d. | n.d. | 109 | 6.6 | 120 | 7.8 |
| BP | n.d. | n.d. | 128 | 4.2 | 90.1 | 2.9 |
| BP3 | n.d. | n.d. | 112 | 6.2 | 122 | 6.6 |
| TCS | 15 ± 1 | 28 ± 2 | 127 | 14 | 121 | 8.7 |
| MBC | 16 ± 1 | n.d. | 97.2 | 2.8 | 82.9 | 2.6 |
| OCR | n.d. | 0.6 ± 0.1 | 95.8 | 29 | 82.2 | 24 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Hernández, P.; Lago, A.B.; Pasán, J.; Ruiz-Pérez, C.; Ayala, J.H.; Afonso, A.M.; Pino, V. Application of a Pillared-Layer Zn-Triazolate Metal-Organic Framework in the Dispersive Miniaturized Solid-Phase Extraction of Personal Care Products from Wastewater Samples. Molecules 2019, 24, 690. https://doi.org/10.3390/molecules24040690
González-Hernández P, Lago AB, Pasán J, Ruiz-Pérez C, Ayala JH, Afonso AM, Pino V. Application of a Pillared-Layer Zn-Triazolate Metal-Organic Framework in the Dispersive Miniaturized Solid-Phase Extraction of Personal Care Products from Wastewater Samples. Molecules. 2019; 24(4):690. https://doi.org/10.3390/molecules24040690
Chicago/Turabian StyleGonzález-Hernández, Providencia, Ana B. Lago, Jorge Pasán, Catalina Ruiz-Pérez, Juan H. Ayala, Ana M. Afonso, and Verónica Pino. 2019. "Application of a Pillared-Layer Zn-Triazolate Metal-Organic Framework in the Dispersive Miniaturized Solid-Phase Extraction of Personal Care Products from Wastewater Samples" Molecules 24, no. 4: 690. https://doi.org/10.3390/molecules24040690
APA StyleGonzález-Hernández, P., Lago, A. B., Pasán, J., Ruiz-Pérez, C., Ayala, J. H., Afonso, A. M., & Pino, V. (2019). Application of a Pillared-Layer Zn-Triazolate Metal-Organic Framework in the Dispersive Miniaturized Solid-Phase Extraction of Personal Care Products from Wastewater Samples. Molecules, 24(4), 690. https://doi.org/10.3390/molecules24040690