
Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
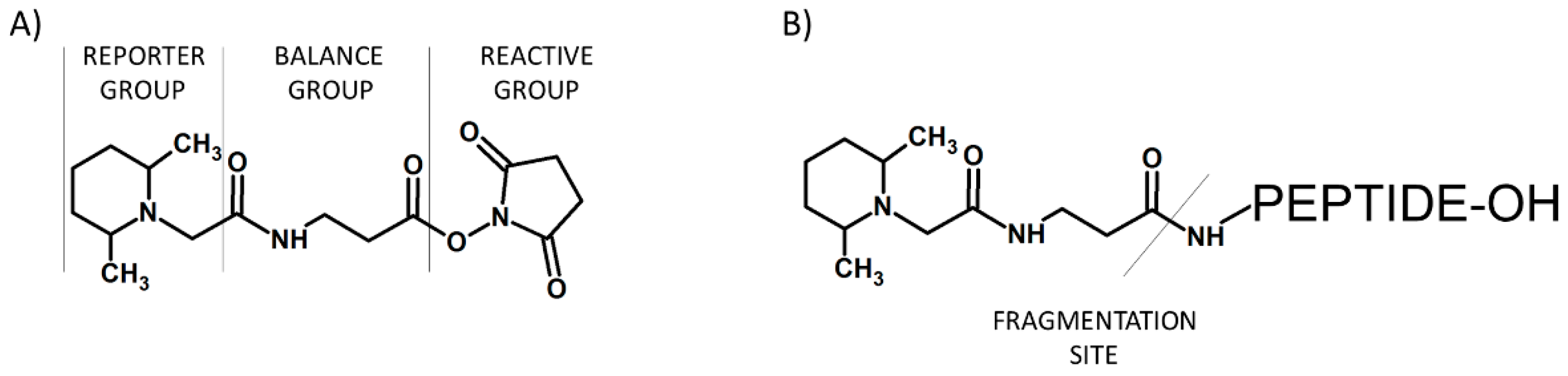{kind=link}
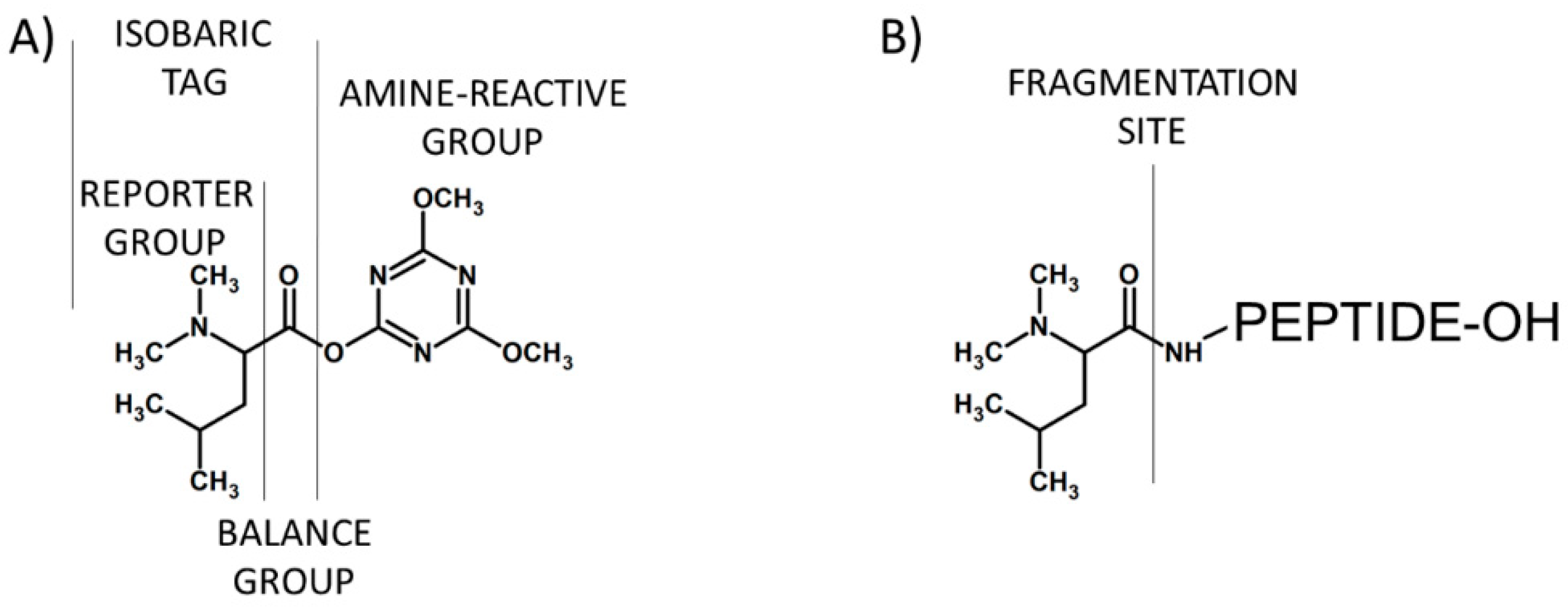{kind=link}
{kind=link}
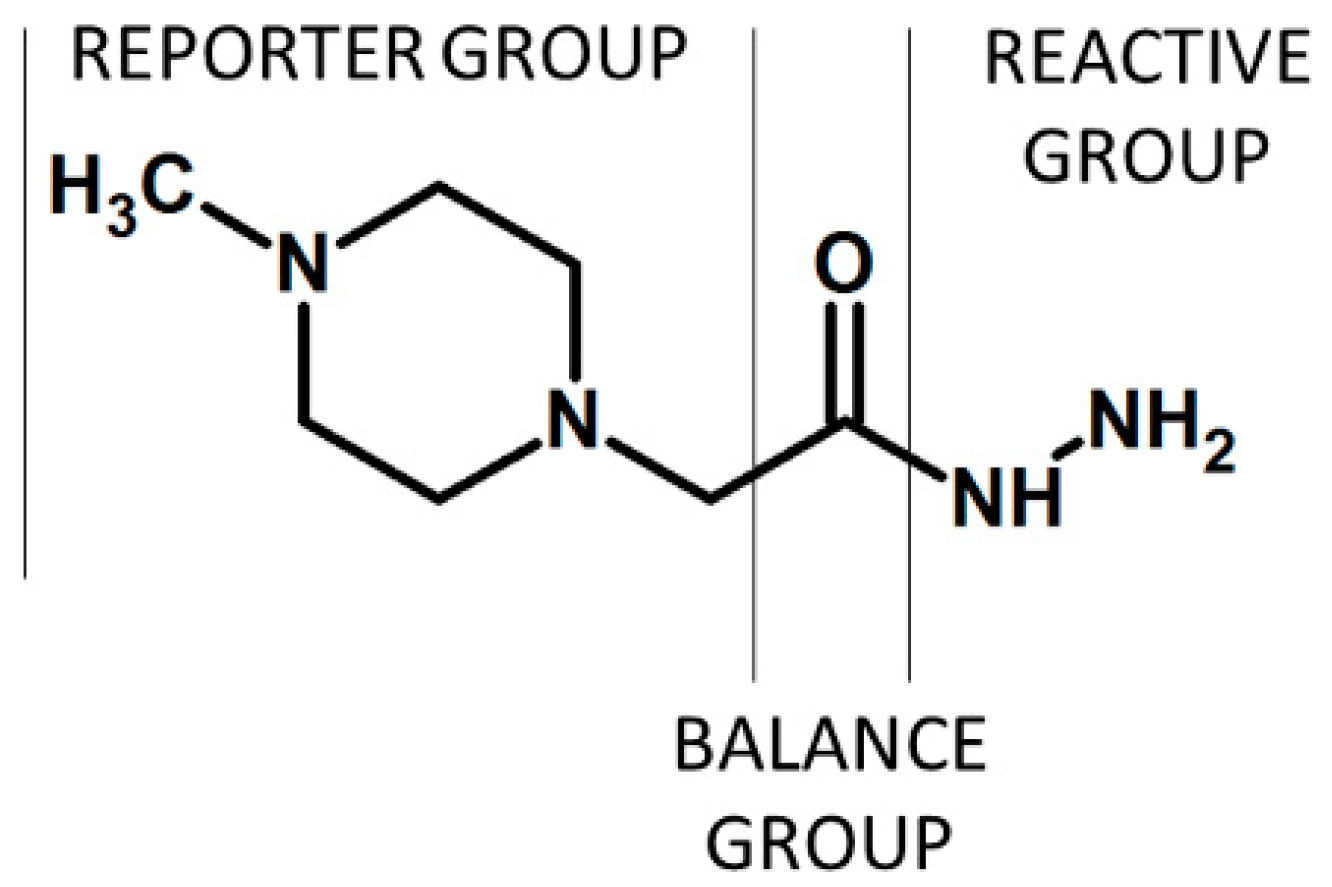{kind=link}
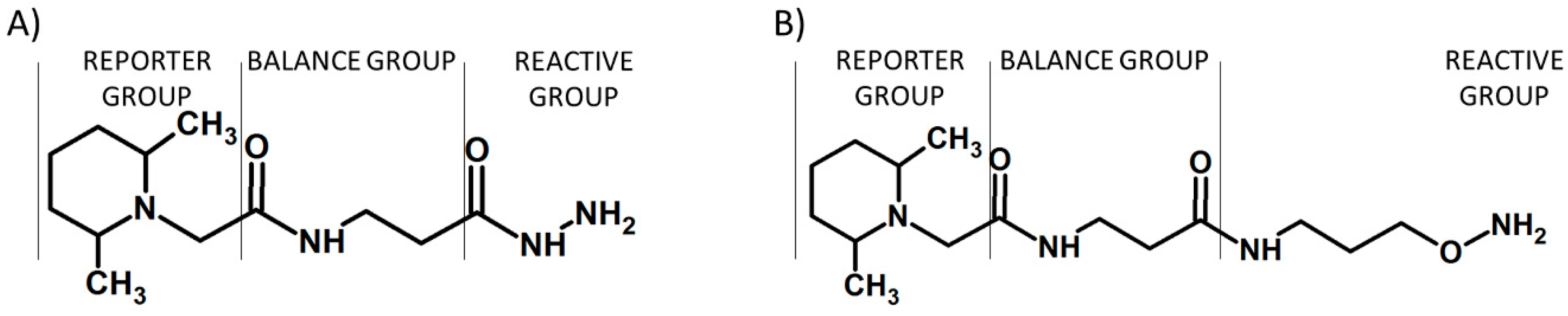{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Increase in sensitivity of detection of the labeled peptides. This goal may be accomplished by incorporating the permanent electric charge to the labeled molecule, which facilitates electrospray ionization (ESI).
- Increase in the multiplexing capacity of isobaric markers. This modification allows to compare bigger number of proteomic samples in a single experiment.
- Modification of reactive group in isobaric tags in order to increase the selectivity and yield of derivatization.
- Simplification of the structure of isobaric tag in order to reduce prices of this group of reagents.
2. ICPL
3. ICAT
4. mTRAQ
5. iTRAQ and TMTs
6. TMT
7. Advantages and Disadvantages
8. DiLeu
9. DiART
Protein Post-Translational Modifications
10. Sensitivity Problem
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collier, T.S.; Sarkar, P.; Franck, W.L.; Rao, B.M.; Dean, R.A.; Muddiman, D.C. Direct comparison of stable isotope labeling by amino acids in cell culture and spectral counting for quantitative proteomics. Anal. Chem. 2010, 82, 8696–8702. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; MacCoss, M.J.; Howell, K.E.; Matthews, D.E.; Yates, J.R., III. Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem. 2004, 76, 4951–4959. [Google Scholar] [CrossRef] [PubMed]
- Bindschedler, L.V.; Palmblad, M.; Cramer, R. Hydroponic isotope labelling of entire plants (HILEP) for quantitative plant proteomics; an oxidative stress case study. Phytochemistry 2008, 69, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.M.; Simpson, D.M.; Doherty, M.K.; Rivers, J.; Gaskell, S.J.; Beynon, R.J. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 2006, 1, 1029–1043. [Google Scholar] [CrossRef]
- Zhang, R.; Regnier, F.E. Minimizing resolution of isotopically coded peptides in comparative proteomics. J. Proteome Res. 2002, 1, 139–147. [Google Scholar] [CrossRef]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 10, 2–20. [Google Scholar] [CrossRef]
- Wiktorowicz, J.E.; English, R.D.; Wu, Z.; Kurosky, A. Model studies on itraq modification of peptides: Sequence-dependent reaction specificity. J. Proteome Res. 2012, 11, 1512–1520. [Google Scholar] [CrossRef]
- Setner, B.; Stefanowicz, P.; Szewczuk, Z. Quaternary ammonium isobaric tag for a relative and absolute quantification of peptides. J. Mass Spectrom. 2018, 53, 115–123. [Google Scholar] [CrossRef]
- Xiang, F.; Ye, H.; Chen, R.; Fu, Q.; Li, L. N,N-Dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 2010, 82, 2817–2825. [Google Scholar] [CrossRef]
- Thomas, J.A.; Mallis, R.J. Aging and oxidation of reactive protein sulfhydryls. Exp. Gerontol. 2001, 36, 1519–1526. [Google Scholar] [CrossRef]
- Palmese, A.; De Rosa, C.; Chiappetta, G.; Marino, G.; Amoresano, A. Novel method to investigate protein carbonylation by iTRAQ strategy. Anal. Bioanal. Chem. 2012, 404, 1631–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, H.; Neubert, P.; Kuhn, K.; Etienne, C.; Bomgarden, R.; Rogers, J.C.; Kuster, B. Carbonyl-reactive tandem mass tags for the proteome-wide quantification of N-linked glycans. Anal. Chem. 2012, 84, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Moulder, R.; Bhosale, S.D.; Goodlett, D.R.; Lahesmaa, R. Analysis of the plasma proteome using iTRAQ and TMT-based Isobaric labeling. Mass Spectrom. Rev. 2018, 37, 583–606. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.H.; Smith, D.S.; Parker, C.E.; Borchers, C. Current trends in quantitative proteomics. J. Mass Spectrom. 2009, 44, 1637–1660. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, Q.K.; Carr, S.A.; Annan, R.S. N-Terminal peptide labeling strategy for incorporation of isotopic tags: A method for the determination of site-specific absolute phosphorylation stoichiometry. Rapid Commun. Mass Spectrom. 2002, 16, 2325–2332. [Google Scholar] [CrossRef]
- Munchbach, M.; Quadroni, M.; Miotto, G.; James, P. Quantitation and facilitated de novo sequencing of proteins by isotopic N-terminal labeling of peptides with a fragmentation-directing moiety. Anal. Chem. 2000, 72, 4047–4057. [Google Scholar] [CrossRef]
- Schmidt, A.; Kellermann, J.; Lottspeich, F. A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics 2005, 5, 4–15. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef]
- Li, J.; Steen, H.; Gygi, S.P. Protein profiling with cleavable isotopecoded affinity tag (cICAT) reagents: The yeast salinity stress response. Mol. Cell. Proteomics 2003, 2, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.C.; Schmitt-Ulms, G.; Chalkley, R.J.; Hirsch, J.; Baldwin, M.A.; Burlingame, A.L. Mass spectrometric analysis of protein mixtures at low levels using cleavable 13C-isotopecoded affinity tag and multidimensional chromatography. Mol. Cell. Proteomics 2003, 2, 299–314. [Google Scholar] [CrossRef]
- Zhang, R.; Sioma, C.S.; Thompson, R.A.; Xiong, L.; Regnier, F.E. Controlling deuterium isotope effects in comparative proteomics. Anal. Chem. 2002, 74, 3662–3669. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Impens, F.; Ghesquiere, B.; Van Damme, P.; Lambrechts, A.; Vandekerckhove, J. Stable isotopic labeling in proteomics. Proteomics 2008, 8, 4873–4885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, A.; Lindner, W. Current chemical tagging strategies for proteome analysis by mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 25, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, C.P.; Crockett, D.K.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Analytical characteristics of cleavable isotope-coded affinity tag- LC-tandem mass spectrometry for quantitative proteomic studies. J. Mol. Diagn. 2006, 8, 513–520. [Google Scholar] [CrossRef]
- Kang, U.-B.; Yeom, J.; Kim, H.; Lee, C. Quantitative analysis of mTRAQ-Labeled proteome using full MS scans. J. Proteome Res. 2010, 9, 3750–3758. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.V.; Taylor, A.M.; Li, W.; Minkoff, M.S.; Romaschin, A.D.; Colgan, T.J.; Siu, K.W. Multiple reaction monitoring of mTRAQ-labeled peptides enables absolute quantification of endogenous levels of a potential cancer marker in cancerous and normal endometrial tissues. J. Proteome Res. 2008, 7, 3525–3534. [Google Scholar] [CrossRef]
- Pichler, P.; Kocher, T.; Holzmann, J.; Mazanek, M.; Taus, T.; Ammerer, G.; Mechtler, K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared toTMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal. Chem. 2010, 82, 6549–6558. [Google Scholar] [CrossRef]
- Pottiez, G.; Wiederin, J.; Fox, H.S.; Ciborowski, P. Comparison of 4-plex to 8-plex iTRAQ Quantitative Measurements of Proteins in Human Plasma Samples. J. Proteome Res. 2012, 11, 3774–3781. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Yates, J.R., III. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteomics 2012, 75, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Everley, R.A.; Kunz, R.C.; McAllister, F.E.; Gygi, S.P. Increasing throughput in targeted proteomics assays: 54-plex quantitation in a single mass spectrometry run. Anal. Chem. 2013, 85, 5340–5346. [Google Scholar] [CrossRef] [PubMed]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [PubMed]
- Sonnett, M.; Yeung, E.; Wühr, M. Accurate, sensitive, and precise multiplexed proteomics using the complement reporter ion cluster. Anal. Chem. 2018, 90, 5032–5039. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; He, Y.; Lin, Z.; Zi, J.; Yang, H.; Zhang, S.; Lou, X.; Wang, Q.; Li, S.; Liu, S. Reagents for Isobaric Labeling Peptides in Quantitative Proteomics. Anal. Chem. 2018, 90, 12366–12371. [Google Scholar] [CrossRef] [PubMed]
- Simpson, K.L.; Whetton, A.D.; Dive, C. Quantitative mass spectrometry-based techniques for clinical use: Biomarker identification and quantification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1240–1249. [Google Scholar] [CrossRef]
- Arul, A.B.; Robinson, R.A.S. Sample Multiplexing Strategies in Quantitative Proteomics. Anal. Chem. 2019, 91, 178–189. [Google Scholar] [CrossRef]
- Frost, D.C.; Greer, T.; Li, L. High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics. Anal. Chem. 2015, 87, 1646–1654. [Google Scholar] [CrossRef]
- Frost, D.C.; Rust, C.J.; Robinson, R.A.S.; Li, L. Increased N,N-Dimethyl Leucine Isobaric Tag Multiplexing by a Combined Precursor Isotopic Labeling and Isobaric Tagging Approach. Anal. Chem. 2018, 90, 10664–10669. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Q.; Lin, L.; Tang, Q.; Edwards, J.L.; Li, S.; Liu, S. Comparative evaluation of two isobaric labeling tags, DiART and iTRAQ. Anal. Chem. 2012, 84, 2908–2915. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Li, S. Deuterium isobaric amine-reactive tags for quantitative proteomics. Anal. Chem. 2010, 82, 7588–7595. [Google Scholar] [CrossRef]
- Leitner, A. A review of the role of chemical modification methods in contemporary mass spectrometry-based proteomics research. Anal. Chim. Acta 2018, 1000, 2–19. [Google Scholar] [CrossRef]
- Lin, D.; Li, J.; Slebos, R.J.; Liebler, D.C. Cysteinyl peptide capture for shotgun proteomics: Global assessment of chemoselective fractionation. J. Proteome Res. 2010, 9, 5461–5472. [Google Scholar] [CrossRef]
- Tambor, V.; Hunter, C.L.; Seymour, S.L.; Kacerovsky, M.; Stulik, J.; Lenco, J. CysTRAQ—A combination of iTRAQ and enrichment of cysteinyl peptides for uncovering and quantifying hidden proteomes. J. Proteomics 2012, 75, 857–867. [Google Scholar] [CrossRef]
- Pan, K.T.; Chen, Y.Y.; Pu, T.H.; Chao, Y.S.; Yang, C.Y.; Bomgarden, R.D.; Rogers, J.C.; Meng, T.C.; Khoo, K.H. Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox Signal. 2014, 20, 1365–1381. [Google Scholar] [CrossRef]
- Qu, Z.; Meng, F.; Bomgarden, R.D.; Viner, R.I.; Li, J.; Rogers, J.C.; Cheng, J.; Greenlief, C.M.; Cui, J.; Lubahn, D.B.; et al. Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 2014, 13, 3200–3211. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, K.; Choe, L.H.; Lee, K.H. Shotgun proteomics using the iTRAQ isobaric tags. Brief. Funct. Genomics Proteomics 2006, 5, 112–120. [Google Scholar] [CrossRef]
- Hardt, M.; Witkowska, H.E.; Webb, S.; Thomas, L.R.; Dixon, S.E.; Hall, S.C.; Fisher, S.J. Assessing the effects of diurnal variation on the composition of human parotid saliva: Quantitative analysis of native peptides using iTRAQ reagents. Anal. Chem. 2005, 77, 4947–4954. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, R.; Setner, B.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. The unusual hydrogen-deuterium exchange of α-carbon protons in N-substituted glycine containing peptides. J. Mass Spectrom. 2014, 49, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, R.; Rudowska, M.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. Hydrogen-deuterium exchange of α-carbon protons and fragmentation pathways in N-methylated glycine and alanine-containing peptides derivatized by quaternary ammonium salts. J. Mass Spectrom. 2014, 49, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, R.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. Facile synthesis of deuterium-labeled denatonium cation and its application in the quantitative analysis of Bitrex by liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 6557–6561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bąchor, R.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. Preparation of novel deuterated cyclosporin A standards for quantitative LC-MS analysis. J. Mass Spectrom. 2017, 52, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Vetter, D.E.; Basappa, J.; Turcan, S. Multiplexed isobaric tagging protocols for quantitative mass spectrometry approaches to auditory research. Methods Mol. Biol. 2009, 493, 345–366. [Google Scholar] [PubMed]
- Protein Quantitation (iTRAQ). Available online: https://www.proteomics.com.au/analytical-services/itraq/ (accessed on 13 December 2018).
- Stults, J.T.; Lai, J.; McCune, S.; Wetzel, R. Simplification of high-energy collision spectra of peptides by amino-terminal derivatization. Anal. Chem. 1993, 65, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Vath, J.E.; Biemann, K. Microderivatization of peptides by placing a fixed positive charge at the N-terminus to modify high-energy collision fragmentation. Int. J. Mass Spectrom. Ion Processes. 1990, 100, 287–299. [Google Scholar] [CrossRef]
- Mirzaei, H.; Regnier, F. Enhancing electrospray ionization efficiency of peptides by derivatization. Anal. Chem. 2006, 78, 4175–4183. [Google Scholar] [CrossRef]
- Liao, P.C.; Huang, Z.H.; Allison, J. Charge remote fragmentation of peptides following attachment of a fixed positive charge. J. Am. Soc. Mass Spectrom. 1997, 8, 501–509. [Google Scholar] [CrossRef]
- Blagojevic, V.; Zhidkov, N.; Tharmaratnam, S.; Pham, V.T.; Kaplan, H.; Bohme, D.K. Peptide quantitation with methyl iodide isotopic tags and mass spectrometry. Analyst 2010, 135, 1456–1460. [Google Scholar] [CrossRef]
- Cydzik, M.; Rudowska, M.; Stefanowicz, P.; Szewczuk, Z. The competition of charge remote and charge directed fragmentation mechanisms in quaternary ammonium salt derivatized peptides—An isotopic exchange study. J. Am. Soc. Mass Spectrom. 2011, 22, 2103–2107. [Google Scholar] [CrossRef]
- Setner, B.; Rudowska, M.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. The 5-azoniaspiro[4.4]nonyl group for improved MS peptide analysis: A novel non-fragmenting ionization tag for mass spectrometric sensitive sequencing of peptides. Anal. Chim. Acta 2017, 986, 71–81. [Google Scholar] [CrossRef]
- Cydzik, M.; Rudowska, M.; Stefanowicz, P.; Szewczuk, Z. Derivatization of peptides as quaternary ammonium salts for sensitive detection by ESI–MS. J. Pept. Sci. 2011, 17, 445–453. [Google Scholar] [CrossRef]
- Bąchor, R.; Mielczarek, P.; Rudowska, M.; Silberring, J.; Szewczul, Z. Sensitive detection of charge peptides at the attomole level using nano-LC-ESI-MRM analysis. Int. J. Mass Spectrom. 2014, 362, 32–38. [Google Scholar]
- Rudowska, M.; Wojewska, D.; Kluczyk, A.; Bąchor, R.; Stefanowicz, P.; Szewczuk, Z. The hydrogen-deuterium exchange at α-carbon atom in N,N,N-trialkylglycine residue: ESI-MS studies. J. Am. Soc. Mass Spectrom. 2012, 23, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Waliczek, M.; Kijewska, M.; Rudowska, M.; Setner, B.; Stefanowicz, P.; Szewczuk, Z. Peptides labeled with pyridinium salts for sensitive detection and sequencing by electrospray tandem mass spectrometry. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Balaban, A.T.; Boulton, A.J. 2,4,6-Trimethyl-pyrylium tetrafluoroborate. Org. Synth. 1973, 5, 1112–1113. [Google Scholar]
- Balaban, A.T.; Boulton, A.J. 2,4,6-Trimethyl-pyrylium trifluoromethanesulfonate. Org. Synth. 1973, 5, 1114–1116. [Google Scholar]
- Dimroth, K.; Reichardt, C.; Vogel, K. 2,4,6-triphenylpyrylium tetrafluoroborate. Org. Synth. 1973, 5, 1135. [Google Scholar]
- Waliczek, M.; Bąchor, R.; Kijewska, M.; Gąszczyk, D.; Panek-Laszczyńska, K.; Dąbrowska, K.; Witkiewicz, W.; Marek-Bukowiec, K.; Tracz, J.; et al. Isobaric duplex based on a combination of 16O/18O enzymatic exchange and labeling with pyrylium salts. Anal. Chim. Acta 2019, 1048, 96–104. [Google Scholar] [CrossRef]
- Yao, X.; Afonso, C.; Fenselau, C. Dissection of proteolytic 18O labeling: Endoprotease-catalyzed 16O-to-18O exchange of truncated peptide substrates. J. Proteome Res. 2003, 2, 147–152. [Google Scholar] [CrossRef]
- Rao, K.C.; Carruth, R.T.; Miyagi, M. Proteolytic 18O labeling by peptidyl-Lys metalloendopeptidase for comparative proteomics. J. Proteome Res. 2005, 4, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Abe, K.; Yamaguchi, H.; Goto, J.; Shimada, M.; Mano, N. Production of 18O-single labeled peptide fragments during trypsin digestion of proteins for quantitaive proteomcis using nanoLC-ESI-MS/MS. J. Proteome Res. 2010, 9, 3741–3749. [Google Scholar] [CrossRef] [PubMed]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bąchor, R.; Waliczek, M.; Stefanowicz, P.; Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules 2019, 24, 701. https://doi.org/10.3390/molecules24040701
Bąchor R, Waliczek M, Stefanowicz P, Szewczuk Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 2019; 24(4):701. https://doi.org/10.3390/molecules24040701
Chicago/Turabian StyleBąchor, Remigiusz, Mateusz Waliczek, Piotr Stefanowicz, and Zbigniew Szewczuk. 2019. "Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics" Molecules 24, no. 4: 701. https://doi.org/10.3390/molecules24040701
APA StyleBąchor, R., Waliczek, M., Stefanowicz, P., & Szewczuk, Z. (2019). Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules, 24(4), 701. https://doi.org/10.3390/molecules24040701