
Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies

 , , , ,
, , , ,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Synthesis of the Oxygenated Xanthones (OXs)
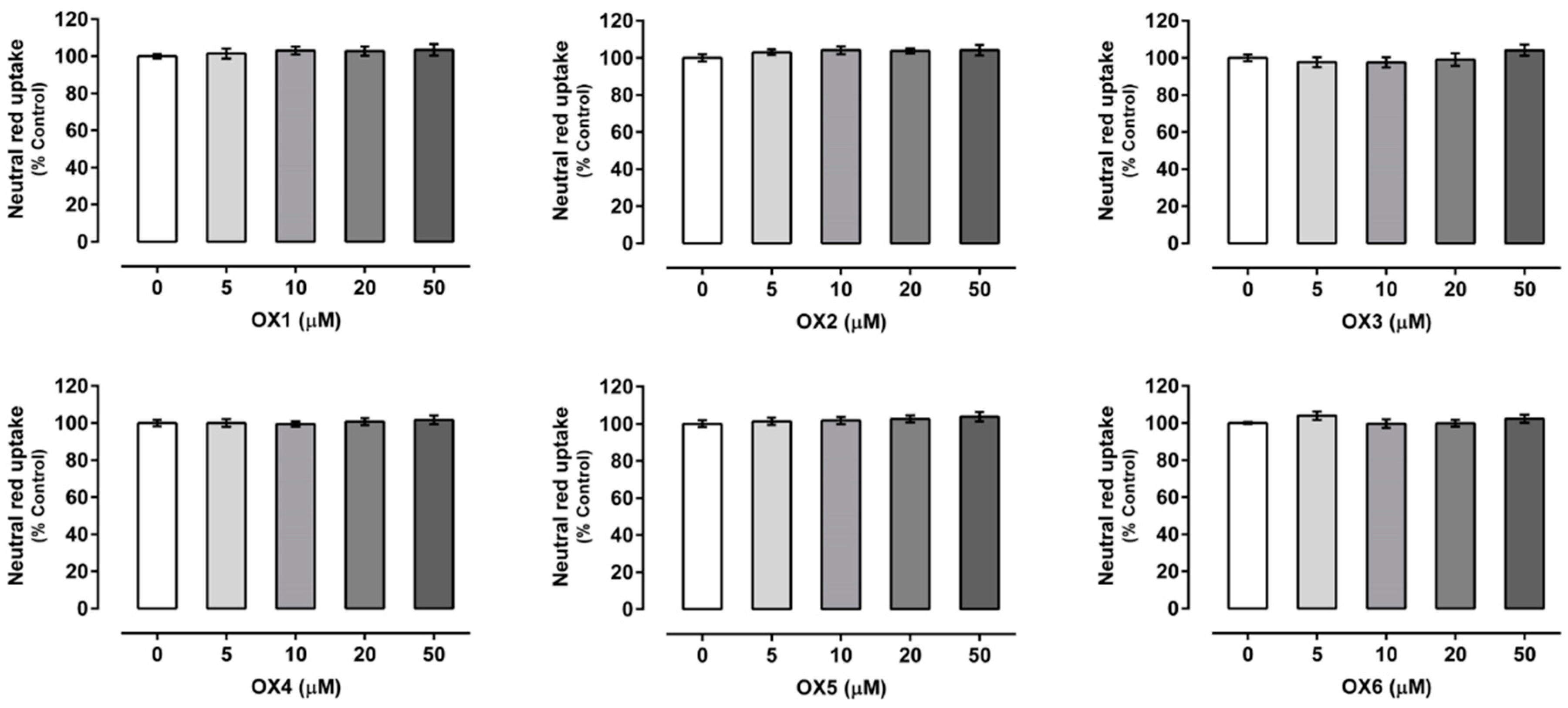
2.2. Oxygenated Xanthones Cytotoxicity Assays
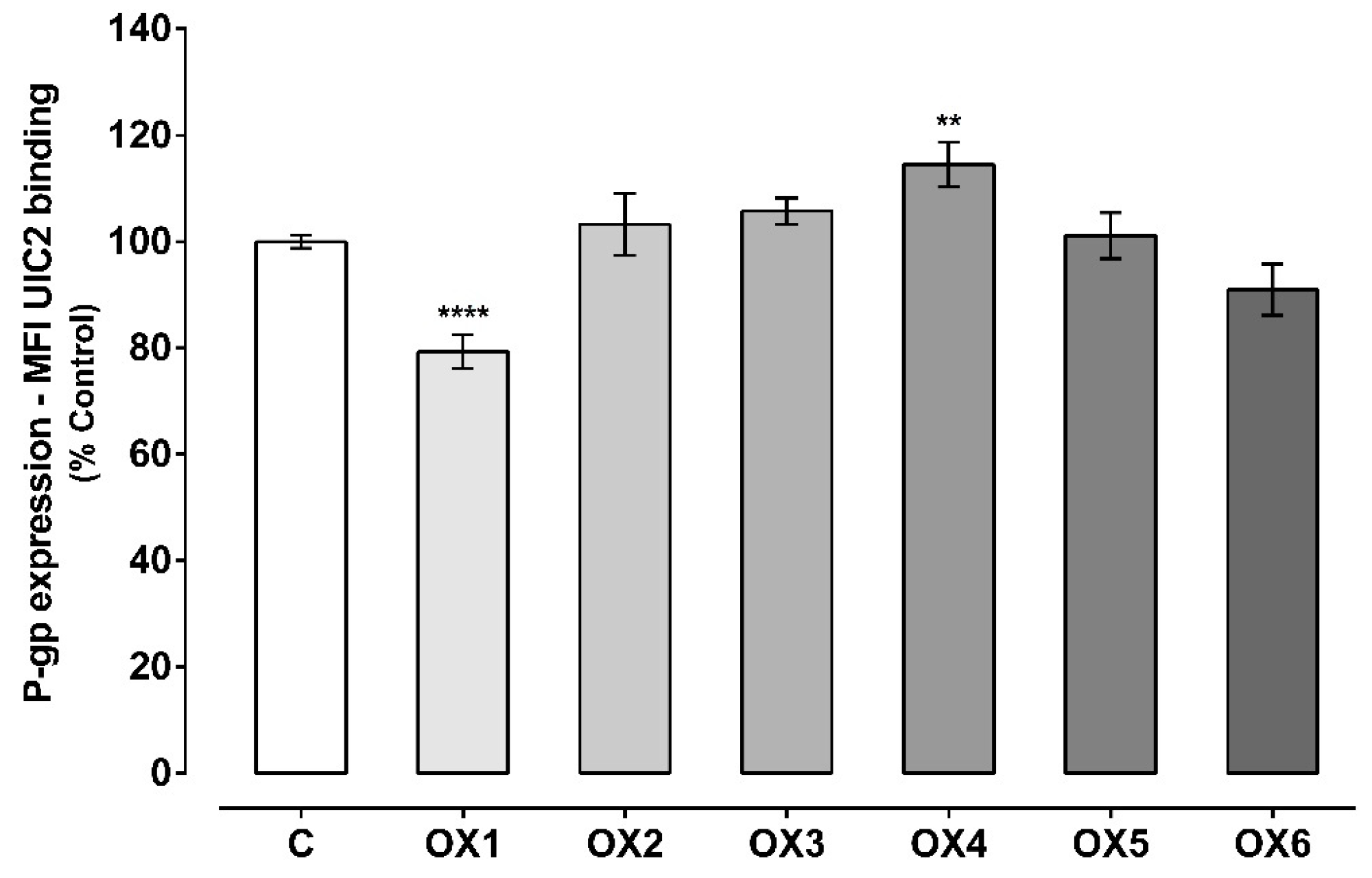
2.3. Evaluation of P-Glycoprotein Expression
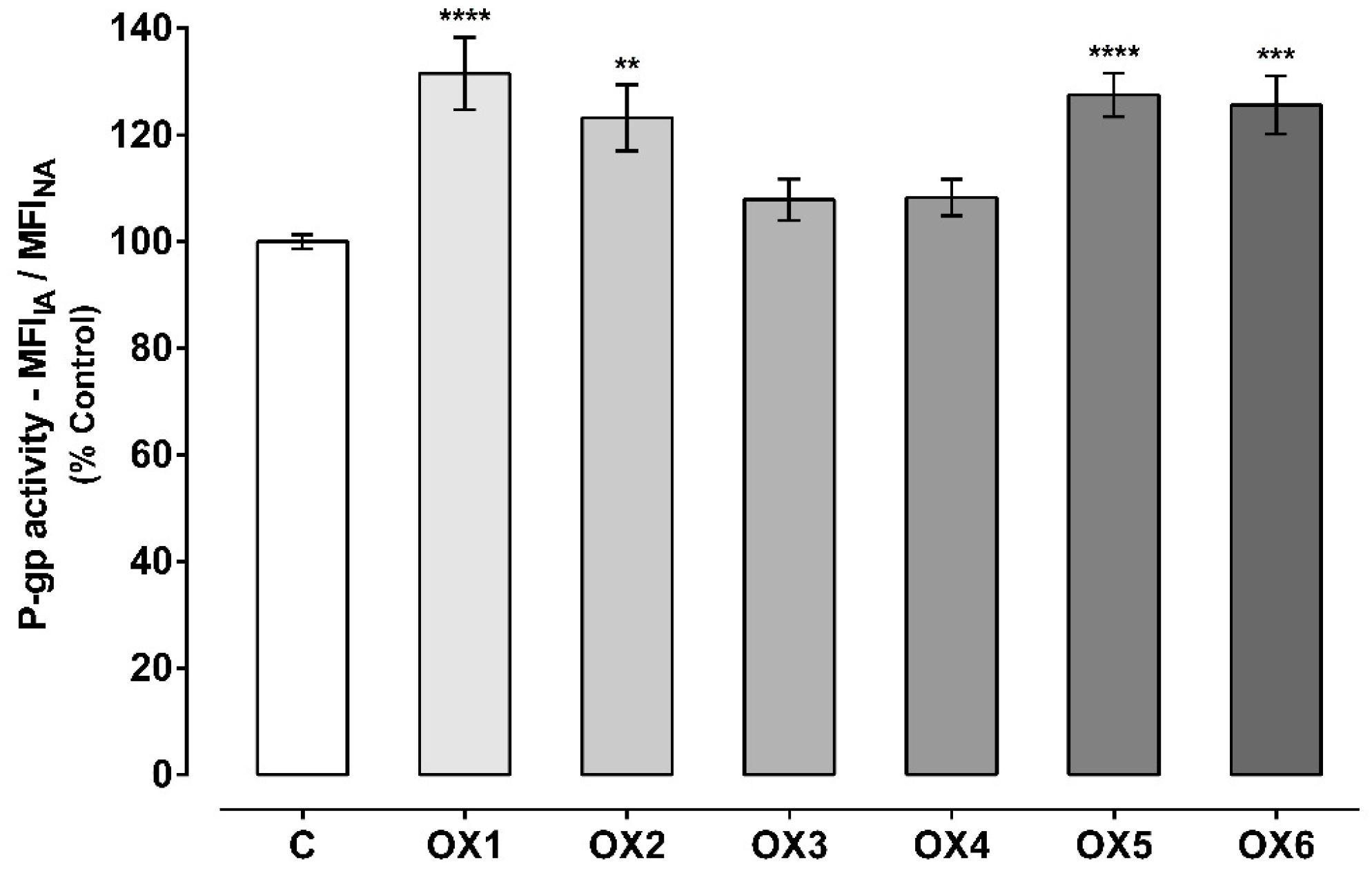
2.4. Evaluation of P-Glycoprotein Transport Activity
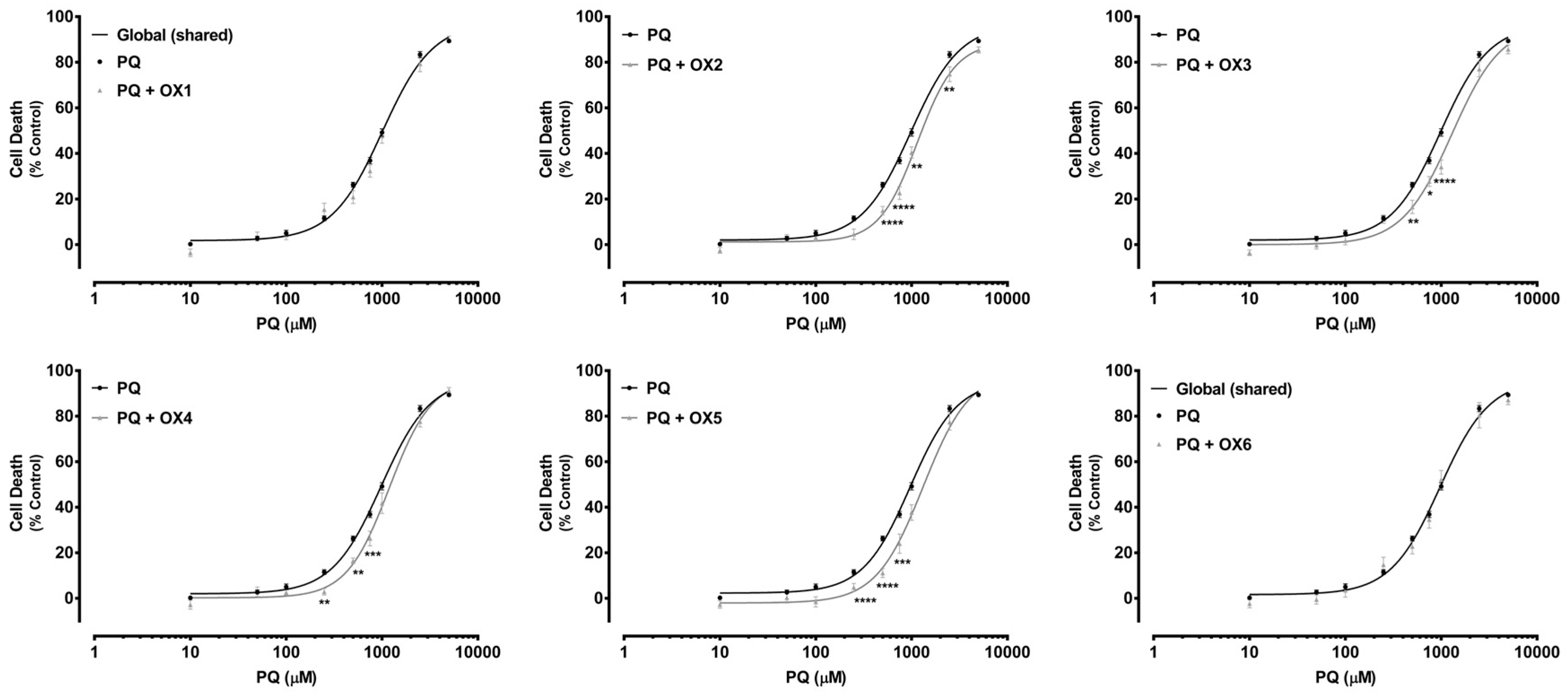
2.5. Oxygenated Xanthones’ Protective Effects Against Paraquat-Induced Cytotoxicity
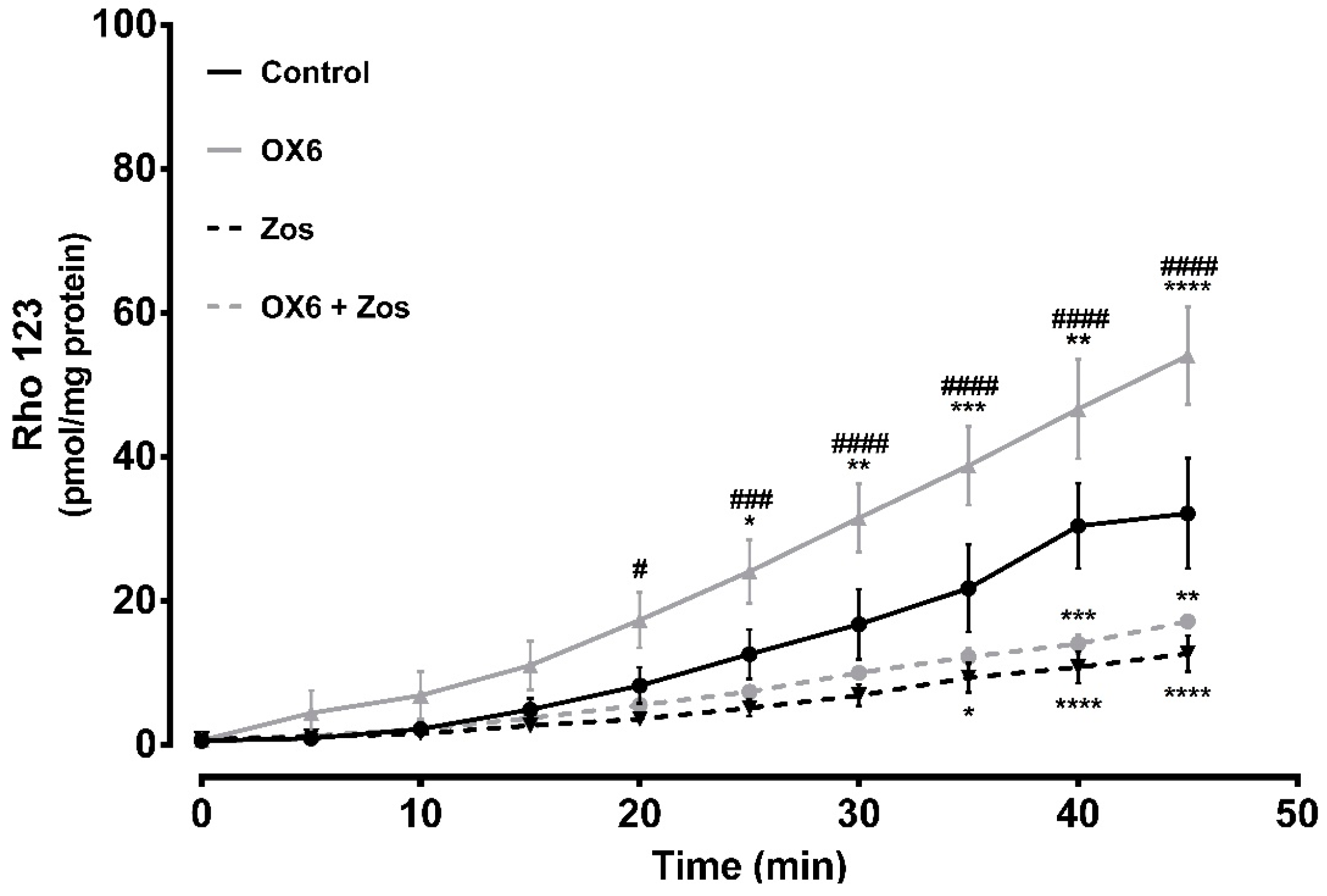
2.6. Ex Vivo Studies—Evaluation of P-Glycoprotein Transport Activity in Rat Everted Intestinal Sacs
2.7. In Silico Studies
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of the Oxygenated Xanthones (OXs)
4.2.1. General Information
4.2.2. Synthesis of 3,4-dimethoxy-1-methyl-9H-xanthen-9-one (OX1)
4.2.3. Synthesis of 1-(dibromomethyl)-3,4-dimethoxy-9H-xanthen-9-one (OX2)
4.2.4. Synthesis of 3,6-dihydroxy-9-oxo-9H-xanthene-4,5-dicarbaldehyde (OX3)
4.2.5. Synthesis of 4-hydroxy-3-methoxy-9-oxo-9H-xanthene-1-carbaldehyde (OX4)
4.2.6. Synthesis of 3,4-dimethoxy-9-oxo-9H-xanthene-1-carbaldehyde (OX5)
4.2.7. Synthesis of 1-(hydroxymethyl)-3,4-dimethoxy-9H-xanthen-9-one (OX6)
4.3. Caco-2 Cell Culture
4.4. Oxygenated Xanthones’ Cytotoxicity
4.4.1. Neutral Red Uptake Assay
4.5. Evaluation of P-Glycoprotein Expression
4.6. Evaluation of P-Glycoprotein Transport Activity
4.6.1. Rhodamine 123 Efflux in the Presence of the Tested Oxygenated Xanthones
4.6.2. Rhodamine 123 Efflux in Cells Pre-Exposed to the Tested Oxygenated Xanthones
4.7. Paraquat Cytotoxic Assays
4.8. Ex Vivo Studies
4.8.1. Animals
4.8.2. Effect of Oxygenated Xanthone OX6 on P-Glycoprotein Activity–Ex Vivo
4.9. Statistical Analysis
4.10. In Silico Studies
4.10.1. P-Glycoprotein Model Construction
4.10.2. Docking Study
4.10.3. Mapping of Small Molecules onto Pharmacophores
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sharom, F.J. Abc multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Palmeira, A.; Carmo, H.; Barbosa, D.J.; Gameiro, M.; Gomes, A.; Paiva, A.M.; Sousa, E.; Pinto, M.; Bastos Mde, L.; et al. P-glycoprotein induction in caco-2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity. Arch. Toxicol. 2015, 89, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, function and regulation of p-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; Bastos Mde, L.; Remiao, F. Induction and activation of p-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the p-glycoprotein substrate paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Spiers, J.P. A primer on the mechanics of p-glycoprotein the multidrug transporter. Pharmacol. Res. 2007, 55, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of p-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product p-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef]
- Arceci, R.J.; Croop, J.M.; Horwitz, S.B.; Housman, D. The gene encoding multidrug resistance is induced and expressed at high levels during pregnancy in the secretory epithelium of the uterus. Proc. Natl. Acad. Sci. USA 1988, 85, 4350–4354. [Google Scholar] [CrossRef]
- Chin, J.E.; Soffir, R.; Noonan, K.E.; Choi, K.; Roninson, I.B. Structure and expression of the human mdr (p-glycoprotein) gene family. Mol. Cell Biol. 1989, 9, 3808–3820. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Boccia, J.; Casals, D.; Bertino, J.R.; Melamed, M.R. Expression of the multidrug resistance gene product (p-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990, 38, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Gil, S.; Saura, R.; Forestier, F.; Farinotti, R. P-glycoprotein expression of the human placenta during pregnancy. Placenta 2005, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Zhang, Z.J.; Tsuzuki, H.; Ohtsubo, T.; Yamada, T.; Yamamoto, T.; Saito, H. Expression of p-glycoprotein in inner ear capillary endothelial cells of the guinea pig with special reference to blood-inner ear barrier. Brain Res. 1997, 767, 388–392. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Sharom, F.J. The p-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. Abc efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Doring, B.; Petzinger, E. Phase 0 and phase iii transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef]
- Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Remiao, F. Cellular models and in vitro assays for the screening of modulators of p-gp, mrp1 and bcrp. Molecules 2017, 22, 600. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; de Pinho, P.G.; Dinis-Oliveira, R.J.; Carvalho, F.; Silva, I.; Correia-de-Sa, P.; Bastos Mde, L.; Remiao, F. Doxorubicin decreases paraquat accumulation and toxicity in caco-2 cells. Toxicol. Lett. 2013, 217, 34–41. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Dinis-Oliveira, R.; Cordeiro-da-Silva, A.; Lima, S.C.; Carvalho, F.; Bastos Mde, L.; Remiao, F. In vitro study of p-glycoprotein induction as an antidotal pathway to prevent cytotoxicity in caco-2 cells. Arch. Toxicol. 2011, 85, 315–326. [Google Scholar] [CrossRef]
- Bisi, A.; Micucci, M.; Gobbi, S.; Belluti, F.; Budriesi, R.; Rampa, A. Cardiovascular profile of xanthone-based 1,4 dihydropyridines bearing a lidoflazine pharmacophore fragment. Molecules 2018, 23, 3088. [Google Scholar] [CrossRef] [PubMed]
- Resende, D.I.S.P.; Pereira-Terra, P.; Inácio, Â.S.; Costa, P.M.d.; Pinto, E.; Sousa, E.; Pinto, M.M.M. Lichen xanthones as models for new antifungal agents. Molecules 2018, 23, 2617. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.; Sousa, M.E.; Nascimento, M.S. Xanthone derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the tap73 interaction with mdm2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; Bastos Mde, L.; Remiao, F. Colchicine effect on p-glycoprotein expression and activity: In silico and in vitro studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Martins, E.; Silva, R.; Pinto, M.M.M.; Remiao, F.; Sousa, E.; Fernandes, C. Chiral thioxanthones as modulators of p-glycoprotein: Synthesis and enantioselectivity studies. Molecules 2018, 23, 626. [Google Scholar] [CrossRef]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; de Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef]
- Quillinan, A.J.; Scheinmann, F. Studies in the xanthone series. Part XII. A general synthesis of polyoxygenated xanthones from benzophenone precursors. J. Chem. Soc. Perk. Trans. 1 1973, 1329–1337. [Google Scholar] [CrossRef]
- Cruz, I.; Puthongking, P.; Cravo, S.; Palmeira, A.; Cidade, H.; Pinto, M.; Sousa, E. Xanthone and flavone derivatives as dual agents with acetylcholinesterase inhibition and antioxidant activity as potential anti-alzheimer agents. J. Chem. 2017, 2017. [Google Scholar] [CrossRef]
- Fernandes, E.G.; Silva, A.M.; Cavaleiro, J.A.; Silva, F.M.; Fernanda, M.; Borges, M.; Pinto, M.M. 1H and 13C NMR spectroscopy of mono-, di-, tri- and tetrasubstituted xanthones. Magn. Reson. Chem. 1998, 36, 305–309. [Google Scholar] [CrossRef]
- Estudante, M.; Morais, J.G.; Soveral, G.; Benet, L.Z. Intestinal drug transporters: An overview. Adv. Drug Deliv. Rev. 2013, 65, 1340–1356. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, H.; Yang, K.; Xu, W.; Tang, W.; Li, X. The structure and functions of p-glycoprotein. Curr. Med. Chem. 2010, 17, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Shedding light on drug transport: Structure and function of the p-glycoprotein multidrug transporter (abcb1). Biochem. Cell Biol. 2006, 84, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in p-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of p-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New uses for old drugs: Pharmacophore-based screening for the discovery of p-glycoprotein inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Silva, R.; Gaio, A.R.; Martins, A.M.; Lima, S.C.; Cordeiro-da-Silva, A.; de Lourdes Bastos, M.; Remiao, F. P-glycoprotein activity in human caucasian male lymphocytes does not follow its increased expression during aging. Cytom. Part. A 2011, 79, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Wongwanakul, R.; Vardhanabhuti, N.; Siripong, P.; Jianmongkol, S. Effects of rhinacanthin-c on function and expression of drug efflux transporters in caco-2 cells. Fitoterapia 2013, 89, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, C.I.; Arias, A.; Novak, A.; Carpini, G.D.; Villanueva, S.; Blazquez, A.G.; Marin, J.J.; Mottino, A.D.; Rubio, M.C. Acetaminophen-induced stimulation of mdr1 expression and activity in rat intestine and in ls 174t human intestinal cell line. Biochem. Pharmacol. 2011, 81, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Carpini, G.D.; Ruiz, M.L.; Luquita, M.G.; Rubio, M.C.; Mottino, A.D.; Ghanem, C.I. Acetaminophen inhibits intestinal p-glycoprotein transport activity. J. Pharm. Sci. 2013, 102, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Godoy, Y.C.; Martinez, S.A.; Ghanem, C.I.; Celuch, S.M. Fructose-induced metabolic syndrome decreases protein expression and activity of intestinal p-glycoprotein. Nutrition 2015, 31, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Borgnia, M.J.; Eytan, G.D.; Assaraf, Y.G. Competition of hydrophobic peptides, cytotoxic drugs, and chemosensitizers on a common p-glycoprotein pharmacophore as revealed by its atpase activity. J. Biol. Chem. 1996, 271, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Ayesh, S.; Shao, Y.M.; Stein, W.D. Co-operative, competitive and non-competitive interactions between modulators of p-glycoprotein. Biochim. Biophys. Acta 1996, 1316, 8–18. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Positively cooperative sites for drug transport by p-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.F.; Callaghan, R.; Modok, S.; Higgins, C.F.; Ford, R.C. Three-dimensional structure of p-glycoprotein: The transmembrane regions adopt an asymmetric configuration in the nucleotide-bound state. J. Biol. Chem. 2005, 280, 2857–2862. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Kamis, A.B.; Callaghan, R.; Higgins, C.F.; Ford, R.C. Three-dimensional structures of the mammalian multidrug resistance p-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J. Biol. Chem. 2003, 278, 8294–8299. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Velarde, G.; Ford, R.C.; Martin, C.; Berridge, G.; Kerr, I.D.; Callaghan, R.; Schmidlin, A.; Wooding, C.; Linton, K.J.; et al. Repacking of the transmembrane domains of p-glycoprotein during the transport atpase cycle. EMBO J. 2001, 20, 5615–5625. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.; Fernandes, M.X. Structure and ligand-based design of p-glycoprotein inhibitors: A historical perspective. Curr. Pharm. Des. 2012, 18, 4197–4214. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of p-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of p-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse p-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human p-glycoprotein in the atp-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Kairys, V.; Gilson, M.K.; Fernandes, M.X. Using protein homology models for structure-based studies: Approaches to model refinement. The Scientific World J. 2006, 6, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Nayeem, A.; Sitkoff, D.; Krystek, S., Jr. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006, 15, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Tarcsay, A.; Keseru, G.M. Homology modeling and binding site assessment of the human p-glycoprotein. Future Med. Chem. 2011, 3, 297–307. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Mapping the binding site of the inhibitor tariquidar that stabilizes the first transmembrane domain of p-glycoprotein. J. Biol. Chem. 2015, 290, 29389–29401. [Google Scholar] [CrossRef]
- Bansal, T.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Emerging significance of flavonoids as p-glycoprotein inhibitors in cancer chemotherapy. J. Pharm. Pharm. Sci. 2009, 12, 46–78. [Google Scholar] [CrossRef]
- Sterz, K.; Mollmann, L.; Jacobs, A.; Baumert, D.; Wiese, M. Activators of p-glycoprotein: Structure-activity relationships and investigation of their mode of action. ChemMedChem 2009, 4, 1897–1911. [Google Scholar] [CrossRef]
- Garrigos, M.; Mir, L.M.; Orlowski, S. Competitive and non-competitive inhibition of the multidrug-resistance-associated p-glycoprotein atpase—Further experimental evidence for a multisite model. Eur. J. Biochem. 1997, 244, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Zeuthen, T.; Skovsgaard, T.; Stein, W.D. Competitive, non-competitive and cooperative interactions between substrates of p-glycoprotein as measured by its atpase activity. Biochim. Biophys. Acta 1997, 1361, 169–176. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Determining the structure and mechanism of the human multidrug resistance p-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim. Biophys. Acta 1999, 1461, 315–325. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Identification of residues within the drug-binding domain of the human multidrug resistance p-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 2000, 275, 39272–39278. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Blundell, T.L.; Mizuguchi, K. Fugue: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001, 310, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Homer, R.W.; Swanson, J.; Jilek, R.J.; Hurst, T.; Clark, R.D. Sybyl line notation (sln): A single notation to represent chemical structures, queries, reactions, and virtual libraries. J. Chem. Inf. Model. 2008, 48, 2294–2307. [Google Scholar] [CrossRef]
- Stebbings, L.A.; Mizuguchi, K. Homstrad: Recent developments of the homologous protein structure alignment database. Nucleic Acids Res. 2004, 32, D203–D207. [Google Scholar] [CrossRef]
- Dolan, M.A.; Noah, J.W.; Hurt, D. Comparison of common homology modeling algorithms: Application of user-defined alignments. Methods Mol Biol 2012, 857, 399–414. [Google Scholar]
- Montalvao, R.W.; Smith, R.E.; Lovell, S.C.; Blundell, T.L. Choral: A differential geometry approach to the prediction of the cores of protein structures. Bioinformatics 2005, 21, 3719–3725. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Oliva, B.; Fiser, A. A supersecondary structure library and search algorithm for modeling loops in protein structures. Nucleic Acids Res. 2006, 34, 2085–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.E.; Lovell, S.C.; Burke, D.F.; Montalvao, R.W.; Blundell, T.L. Andante: Reducing side-chain rotamer search space during comparative modeling using environment-specific substitution probabilities. Bioinformatics 2007, 23, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Carrascoza, F.; Zaric, S.; Silaghi-Dumitrescu, R. Computational study of protein secondary structure elements: Ramachandran plots revisited. J. Mol. Graph. Model. 2014, 50, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Froimowitz, M. Hyperchem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar]
- Zhang, L.; Zhou, W.; Li, D.H. A descent modified polak–ribière–polyak conjugate gradient method and its global convergence. IMA J. Numer. Anal. 2006, 26, 629–640. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with pymol and autodock/vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using pymol. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PQ | PQ + OX1 | PQ + OX2 | PQ + OX3 | PQ + OX4 | PQ + OX5 | PQ + OX6 | |
|---|---|---|---|---|---|---|---|
| EC50 [half-maximum-effect concentrations, µM (95% CI)] | 982.4 (912.4–1058) | 1124 (900.8–1402) | 1153 * (1013–1313) | 1291 ** (1050–1587) | 1188 * (1027–1375) | 1217 ** (1046–1417) | 943.0 (757.2–1174) |
| Top (maximal cell death, % control) | 96.65 | ~100.0 | 88.76 | 96.80 | 96.48 | 94.89 | 94.51 |
| Bottom (baseline, % control) | 2.008 | 0.9805 | 1.236 | 0.01597 | 0.2445 | −1.129 | −0.2018 |
| Hill slope | 1.694 | 1.524 | 2.178 | 1.674 | 1.955 | 2.088 | 1.618 |
| Curve p value (Comparison between the fitted curves) | - | 0.2139 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.6121 |
| PQ + Ela | PQ + OX1 + Ela | PQ + OX2 + Ela | PQ + OX3 + Ela | PQ + OX4 + Ela | PQ + OX5 + Ela | PQ + OX6 + Ela | |
|---|---|---|---|---|---|---|---|
| EC50 [half-maximum-effect concentrations, µM (95% CI)] | 450.3 (397.4–510.4) | 362.5 (278.8–471.4) | 430.6 (403.5–525.4) | 418.6 (324.3–540.3) | 369.2 (260.3–523.5) | 334.7 (243.5–460.0) | 397.8 (293.0–540.0) |
| Top (maximal cell death, % control) | 97.76 | 94.80 | ~100.0 | 96.10 | 97.37 | 97.84 | ~100.0 |
| Bottom (baseline, % control) | −0.2621 | 2.862 | −4.114 | 2.252 | 5.335 | 2.986 | −2.547 |
| Hill slope | 1.039 | 1.086 | 0.7837 | 1.105 | 0.9732 | 0.8449 | 0.8964 |
| Curve p value (Comparison between the fitted curves) | - | 0.0092 | 0.0591 | 0.5798 | <0.0001 | <0.0001 | 0.2742 |
| A | B | ||||
|---|---|---|---|---|---|
| Ligand | Free Energy of Ligand: P-gp TMD Complex (Kcal.mol−2) | Ligand | Free Energy of Ligand: P-gp NBD Complex (Kcal.mol−2) | ||
| OX1 | −6.5 | OX1 | −6 | ||
| OX2 | −7 | OX2 | −6.2 | ||
| OX3 | −6.7 | OX3 | −5.9 | ||
| OX4 | −6.7 | OX4 | −5.8 | ||
| OX5 | −6.7 | OX5 | −5.8 | ||
| OX6 | −6.7 | OX6 | −6.1 | ||
| P-gp TMD inhibitors (controls) | Verapamil | −6 | P-gp NBD inhibitors (controls) | Tangeretin | −6 |
| Tamoxifen | −7.3 | Baicalein | −6.2 | ||
| Quinidine | −7.7 | Nobiletin | −5.8 | ||
| Dexniguldipine | −7.2 | Sinensetin | −5.7 | ||
| Biricodar | −7 | Quercetin | −5.9 | ||
| SR33557 | −6.5 | 2′,4′-Dihydroxy-6′-methoxy-3′,5′-dimethylchalcone | −6.1 | ||
| Elacridar | −9.1 | 3,3′,4′,5,6,7,8-Heptamethoxyflavone | −6 | ||
| Zosuquidar | −8.6 | 3′,4′,7-Trimethoxyflavone | −5.9 | ||
| Tariquidar | −8.4 | ||||
| P-gp activators (controls) | Indirubin | −7.6 | |||
| Coelenteramide | −8.7 | ||||
| Blebbistatin | −8.8 | ||||
| Lefluromide | −7.7 | ||||
| Fit Values | ||||
|---|---|---|---|---|
| Compound | Pharmacophore I (6 features)  | Pharmacophore III (5 features)  | Pharmacophore IV (3 features)  | Pharmacophore V (3 features)  |
| OX1 | 0.485713 | No match | 1.99999 | 2.40877 |
| OX2 | 1.01167 | No match | 2 | 2.91681 |
| OX3 | 2.14728 | 1.77954 | 1.98371 | 2.98381 |
| OX4 | 0.697596 | 1.7693 | 1.84491 | 2.99583 |
| OX5 | 1.2425 | 1.50716 | 1.99911 | 2.81259 |
| OX6 | 1.37048 | 1.84221 | 2.91711 | 2.62604 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, E.; Silva, V.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; Rocha-Pereira, C.; Ghanem, C.I.; Carmo, H.; Remião, F.; et al. Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies. Molecules 2019, 24, 707. https://doi.org/10.3390/molecules24040707
Martins E, Silva V, Lemos A, Palmeira A, Puthongking P, Sousa E, Rocha-Pereira C, Ghanem CI, Carmo H, Remião F, et al. Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies. Molecules. 2019; 24(4):707. https://doi.org/10.3390/molecules24040707
Chicago/Turabian StyleMartins, Eva, Vera Silva, Agostinho Lemos, Andreia Palmeira, Ploenthip Puthongking, Emília Sousa, Carolina Rocha-Pereira, Carolina I. Ghanem, Helena Carmo, Fernando Remião, and et al. 2019. "Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies" Molecules 24, no. 4: 707. https://doi.org/10.3390/molecules24040707
APA StyleMartins, E., Silva, V., Lemos, A., Palmeira, A., Puthongking, P., Sousa, E., Rocha-Pereira, C., Ghanem, C. I., Carmo, H., Remião, F., & Silva, R. (2019). Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies. Molecules, 24(4), 707. https://doi.org/10.3390/molecules24040707