3.1. Chemistry
All commercially available solvents and reagents were used without further purification. Melting points were uncorrected. NMR experiments were performed on Bruker Avance III 400 MHz and Bruker Fourier 300 MHz (Bruker Corporation, Billerica, MA, USA). The spectra were referenced internally according to residual solvent signals of TMS (δ = 0.00 ppm). Positive or negative ion LCMS data were obtained at 303 K by a quadrupole mass spectrometer Agilent LC/MSD 1200 Series (Agilent technologies Inc, Palo Alto, CA, USA) using a 50 × 4.6 mm (5 μm) ODS column. Reversed-phase-HPLC experiments were performed by flash welchrom C18 column (150 × 20 mm) chromatography (Agela Technologies, Tianjin, China). The NMR spectrum of compounds were shown in the
Supplementary Flies (Figures S1–S11).
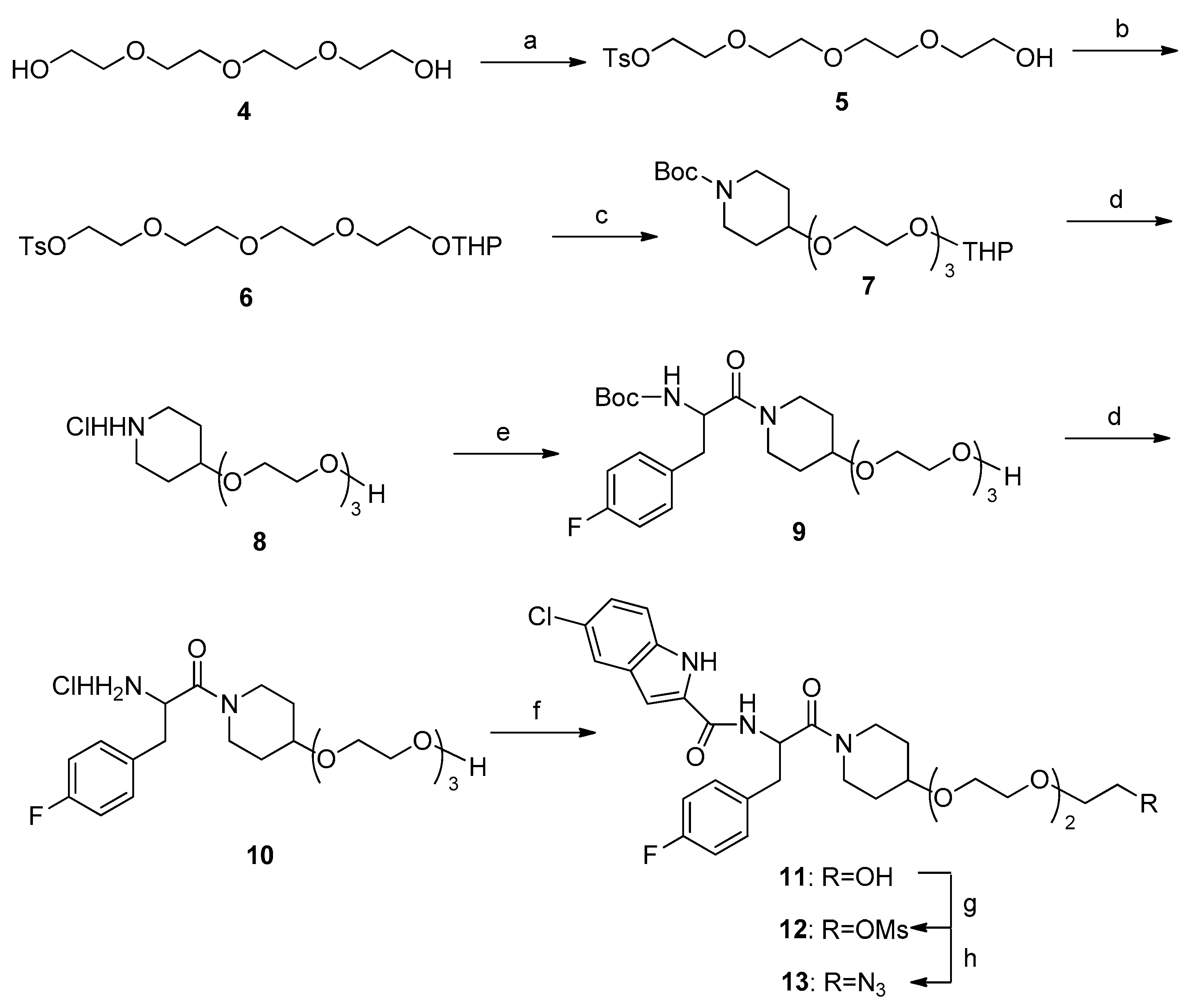
3.1.1. Ethyl 2-{2-[2-(2-hydroxyethoxy)ethoxy]ethoxy}-4-methylbenzenesulfonate (5)
Under an ice bath, TsCl (40 mL, 7.93 g, 0.042 mol) was slowly added in groups to a mixture of tetraethylene glycol (9.0 mL, 0.05 mol) and Et3N (18.05 mL, 0.13 mol) in anhydrous CH2Cl2 (60 mL). The reaction mixture was stirred at 0 °C overnight. Then, the reaction mixture was diluted with CH2Cl2 and washed with water, 1 M aqueous HCl, and saturated NaHCO3 and brine, and then dried and evaporated over anhydrous Na2SO4. The residue was purified by column chromatography on silica gel [EtOAc-petroleum ether (35–90%)] to give the product (8.77 g, 60%) of colorless oil. HPLC analysis was as follows: 99.0%; 1H-NMR (CDCl3, 400 MHz) δ 7.80 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.17 (t, J = 4.8 Hz, 1H), 3.72–3.68 (m, 4H), 3.66–3.62 (m, 4H), 3.61–3.58 (m, 6H), 2.64 (s, 2H), and 2.45 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ 144.9, 132.9, 129.8, 128.0, 72.5, 70.7, 70.6, 70.4, 70.3, 69.3, 68.7, 61.7, and 21.6.
3.1.2. Ethyl 2-[2-(2-{2-[(tetrahydro-2H-pyran-2-yl)oxy]ethoxy}ethoxy)ethoxy]4-methylbenzenesulfonate (6)
A mixture of Pyridinium 4-toluenesulfonate (PPTS, 0.73 g, 2.90 mmol) and 3,4-2H-dihydropyran (3.93 mL, 43.11 mmol) in CH2Cl2 (50 mL) was added to a solution of compound 5 (10.0 g, 28.74 mmol) in anhydrous CH2Cl2 (50 mL). The reaction mixture was stirred at room temperature for 5 h. Then, the reaction mixture was diluted with CH2Cl2 and washed with water and saturated brine, dried over anhydrous Na2SO4, and evaporated. The residue was purified by column chromatography on silica gel [EtOAc-petroleum ether (15–50%)] to give the product (8.83 g, 71%) of colorless oil. HPLC analysis was as follows: 90.1%; 1H-NMR (CDCl3, 400 MHz) δ 7.80 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.62 (t, J = 3.2 Hz, 1H), 4.18–4.15 (m, 2H), 3.87–3.83 (m, 1H), 3.72–3.47 (m, 15H), 2.45 (s, 3H), 1.86–1.78 (m, 1H), 1.75–1.68 (m, 1H), and 1.63–1.49 (m, 4H); 13C-NMR (CDCl3, 100 MHz) δ 144.8, 133.0, 129.8, 128.0, 99.0, 70.7, 70.64, 70.58, 70.53, 69.2, 68.7, 66.6, 62.2, 30.6, 25.4, 21.6, and 19.5.
3.1.3. Tert-butyl 4-{2-[2-(2-{2-[(tetrahydro-2H-pyran-2-yl)oxy]ethoxy}ethoxy)ethoxy]ethoxy}piperidine-1-carboxylate (7)
A mixture of tert-butyl 4-hydroxypiperidine-1-carboxylate (0.50 g, 2.49 mmol) and NaH (0.3 g, 60%, 7.47 mmol) in THF (35.0 mL) was stirred at 0 °C for 1 h. Then, compound 6 (1.10 g, 2.55 mmol) was slowly added to the reaction, which was stirred at room temperature overnight. The mixture was diluted with water (30 mL) and extracted with EtOAc (30 mL × 3). The combined organic phase was washed with brine, dried over anhydrous Na2SO4, and evaporated. The residue was purified by column chromatography on silica gel [EtOAc-petroleum ether (30–70%)] to give the product (0.58 g, 51%) of colorless oil. HPLC analysis was as follows: 96.8%; 1H-NMR (400 MHz, CDCl3) δ 4.63 (t, J = 3.2 Hz, 1H), 3.90–3.84 (m, 2H), 3.79–3.76 (m, 2H), 3.69–3.63 (m, 15H), 3.51–3.45 (m, 2H), 3.09–3.02 (m, 2H), 1.85–1.81 (m, 3H), 1.76–1.68 (m, 1H), 1.63–1.49 (m, 6H), and 1.45 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 154.8, 98.9, 79.4, 75.1, 70.8, 70.7, 70.6, 70.5, 67.4, 66.6, 62.2, 31.0, 30.6, 28.4, 25.4, and 19.5.
3.1.4. N-{(S)-1-[4-(2-{2-[2-(2-mesyloxyethoxy)ethoxy]ethoxy}ethoxy)piperidin-1-yl]-3-(4-fluorophenyl)-1-oxopropan-2-yl}-5-chloro-1H-indole-2-carboxamide (12)
Under an ice bath, methanesulfonyl chloride (0.02 mL, 0.25 mmol) was slowly added in groups to a mixture of compound 11 (0.15 g, 0.24 mmol) and Et3N (0.10 mL, 0.72 mmol) in anhydrous CH2Cl2 (25 mL). The reaction mixture was stirred at room temperature for 5 h. Then, the reaction mixture was diluted with CH2Cl2 and washed with water, 1 M aqueous HCl, and saturated NaHCO3 and brine, and then dried and evaporated over anhydrous Na2SO4. The residue was purified by column chromatography on silica gel [CH3OH-CH2Cl2 (0–2%)] to give the product (0.15 g, 89%) as a white solid. HPLC analysis was as follows: 100.0%; m.p. 130–132 °C. 1H-NMR (400 MHz, CDCl3) δ 9.64 (d, J = 15.2 Hz, 1H), 7.58 (s, 1H), 7.52 (t, J = 6.8 Hz, 1H), 7.31 (t, J = 6.0 Hz, 1H), 7.21 (d, J = 6.8 Hz, 1H), 7.16–7.14 (m, 2H), 6.95 (t, J = 6.8 Hz, 2H), 6.91–6.86 (m, 1H), 5.37–5.33 (m, 1H), 4.38–4.35 (m, 2H), 3.84–3.74 (m, 3H), 3.66–3.59 (m, 14H), 3.49–3.25 (m, 2H), 3.15–3.05 (m, 5H), 1.83–1.50 (m, 3H), and 1.34–1.29 (m, 1H); 13C-NMR (CDCl3, 100 MHz) δ 169.2, 162.0 (d, J = 195.0 Hz), 160.5, 134.7, 131.8 (d, J = 15.4 Hz), 131.5, 131.1, 131.0, 130.9, 128.6, 126.2, 125.0, 121.2, 115.5 (d, J = 3.4 Hz), 115.3 (d, J = 3.6 Hz), 113.0 (d, J = 3.1 Hz), 73.5, 70.82, 70.76, 70.68, 70.5, 69.2, 69.0, 67.6, 50.0 (d, J = 7.7 Hz), 42.7, 39.3, 38.76, 38.47, 31.0 (d, J = 7.6 Hz), and 30.4 (d, J = 3.6 Hz); EIMS m/z 698.1 [M]+.
3.1.5. General Procedure for Synthesis of Compounds 8 and 10
Aqueous HCl (3 M, 2 mL) was slowly added to a solution of compound 7 or 9 (0.58 g, 1.26 mmol) in methanol (15 mL) in an ice bath. The reaction mixture was stirred at room temperature for 6 h, and then concentrated to give the crude target products 8 or 10, which were used for the next reaction without further purification.
3.1.6. General Procedure for Synthesis of Compounds 9 and 11
HATU (1.0 equiv., dissolved in 1.5 mL DMF) and DIPEA (3.0 equiv., dissolved in 1.5 mL DMF) were added to a solution of N-(tert-Butoxycarbonyl)-4-fluoro-l-phenylalanine or 5-chloroindole-2-carboxylicacid (1.0 equiv.) in anhydrous DMF (1.5 mL). The reaction mixture was stirred at room temperature for 10 min. Then, compound 8 or 10 (1.0 equiv.) was added to the reaction. The mixture was stirred at 45 °C for 5 h. After cooling to room temperature, the mixture was diluted with water (30 mL) and extracted with EtOAc (30 mL × 3). The combined organic phase was washed with brine (1 L × 2), dried over anhydrous Na2SO4, and evaporated. The residue was purified by reversed-phase chromatography (methanol-water (20–90%)) to give the product 9 or 11.
(S)-3-(4-Fluorophenyl)-1-{4-[2-(2-{2-[2-(tetrahydro-2H-pyran-2-yloxy)ethoxy]ethoxy}ethoxy)ethoxy]piperidin-1-yl}-2-(tert-butoxycarbonylamino)propan-1-one (9). HPLC analysis was as follows: 98.9%; 1H-NMR (400 MHz, CDCl3) δ 7.16–7.12 (m, 2H), 6.98–6.94 (m, 2H), 5.39 (t, J = 6.4 Hz, 1H), 4.84–4.79 (m, 1H), 3.81–3.71 (m, 3H), 3.67–3.58 (m, 14H), 3.54–3.18 (m, 4H), 2.98–2.92 (m, 2H), 1.79–1.48 (m, 3H), 1.41 (d, J = 3.6 Hz, 9H), and 1.35–1.26 (m, 1H); 13C-NMR (CDCl3, 100 MHz) δ 169.6, 161.9 (d, J = 194 Hz), 155.0, 132.3 (d, J = 18.4 Hz), 131.0 (d, J = 5.9 Hz), 130.9 (d, J = 6.0 Hz), 115.3 (d, J = 5.3 Hz), 115.1 (d, J = 5.2 Hz), 79.7, 73.7 (d, J = 5.2 Hz), 72.5, 70.81, 70.77, 70.65, 70.61, 70.3, 67.5, 61.7, 50.9, 42.6, 39.4, 39.1, 31.1(d, J = 3.8 Hz), 30.4, and 28.3.
N-{(S)-1-[4-(2-{2-[2-(2-Hydroxyethoxy)ethoxy]ethoxy}ethoxy)piperidin-1-yl]-3-(4-fluorophenyl)-1-oxopropan-2-yl}-5-chloro-1H-indole-2-carboxamide (11). HPLC analysis was as follows: 94.5%; m.p. 103–105 °C. 1H-NMR (400 MHz, CDCl3) δ 11.70 (t, J = 8.4 Hz, 1H), 8.91–8.86 (m, 1H), 7.70 (s, 1H), 7.40 (d, J = 6.8 Hz, 1H), 7.34 (dd, J = 6.4, 4.4 Hz, 2H), 7.25 (s, 1H), 7.17 (dd, J = 6.8, 1.6 Hz, 1H), 7.05 (t, J = 6.8 Hz, 2H), 5.15–5.10 (m, 1H), 4.55–4.49 (m, 1H), 3.98–3.79 (m, 1H), 3.73–3.65 (m, 1H), 3.52–3.48 (m, 14H), 3.42–3.39 (m, 2H), 3.33–3.21 (m, 1H), 3.14–2.94 (m, 3H), 1.75–1.66 (m, 2H), and 1.39–1.19 (m, 2H); 13C-NMR (CDCl3, 100 MHz) δ 169.4, 161.5 (d, J = 190.3 Hz), 160.6, 135.3, 134.4 (d, J = 7.5 Hz), 132.9, 131.7, 131.6, 128.5, 124.7, 124.0, 121.1, 115.3, 115.2, 114.3, 103.4, 74.6, 73.8, 72.8, 70.5, 70.3, 70.2, 67.2 (d, J = 6.1 Hz), 60.7, 50.6 (d, J = 9.6 Hz), 43.1, 42.7, 37.0 (d, J = 8.8 Hz), 31.7 (d, J = 33.1 Hz), and 31.1 (d, J = 41.3 Hz). EIMS m/z 620.1 [M]+.
3.1.7. General Procedure for Synthesis of Compounds 3 and 13
NaN3 (3.0 equiv.) was added to a solution of compound 12 or 20 (1.0 equiv.) in anhydrous DMF (10 mL). The reaction mixture was stirred at 60 °C overnight. Then, the reaction mixture was poured into ice water and extracted with EtOAc (30 mL × 3). The combined organic phase was washed with brine and dried over anhydrous Na2SO4. The residue was purified by column chromatography on silica gel [CH3OH-CH2Cl2 (0–2%)] to give the product (0.10 g, 70%) as a thick solid.
N-{(S)-1-[4-(2-{2-[2-(2-Azidoethoxy)ethoxy]ethoxy}ethoxy)piperidin-1-yl]-3-(4-fluorophenyl)-1-oxopropan-2-yl}-5-chloro-1H-indole-2-carboxamide (13). HPLC analysis was as follows: 96.7%; m.p. 93–95 °C; 1H-NMR (400 MHz, d6-DMSO) δ 11.71 (d, J = 6.8 Hz, 1H), 8.91–8.86 (m, 1H), 7.70 (s, 1H), 7.33 (t, J = 5.6 Hz, 2H), 7.25(s, 1H), 7.17 (d, J =7.2 Hz, 1H), 7.05 (t, J = 6.8 Hz, 2H), 5.17–5.09 (m, 1H), 3.98–3.79 (m, 1H), 3.73–3.65 (m, 1H), 3.59–3.48 (m, 15H), 3.41–3.34 (m, 2H), 3.25–2.94 (m, 4H), 1.78–1.65 (m, 2H), and 1.39–1.20 (m, 2H); 13C-NMR (d6-DMSO, 100 MHz) δ 169.3, 161.4 (d, J = 192.1 Hz), 160.6, 135.3, 134.4, 132.9, 131.7, 131.6, 128.5, 124.7, 124.0, 121.1, 115.3 (d, J = 2.5 Hz), 115.1 (d, J = 2.6 Hz), 114.3, 103.4 (d, J = 3.9 Hz), 74.6, 73.8, 70.5, 70.3, 70.2, 70.1, 70.0, 67.2 (d, J = 6.4 Hz), 50.6 (d, J = 8.4 Hz), 50.5, 43.1, 42.7, 37.0 (d, J = 6.8 Hz), 31.7 (d, J = 33 Hz), and 31.1 (d, J = 40.1 Hz). EIMS m/z 644.8 [M]+.
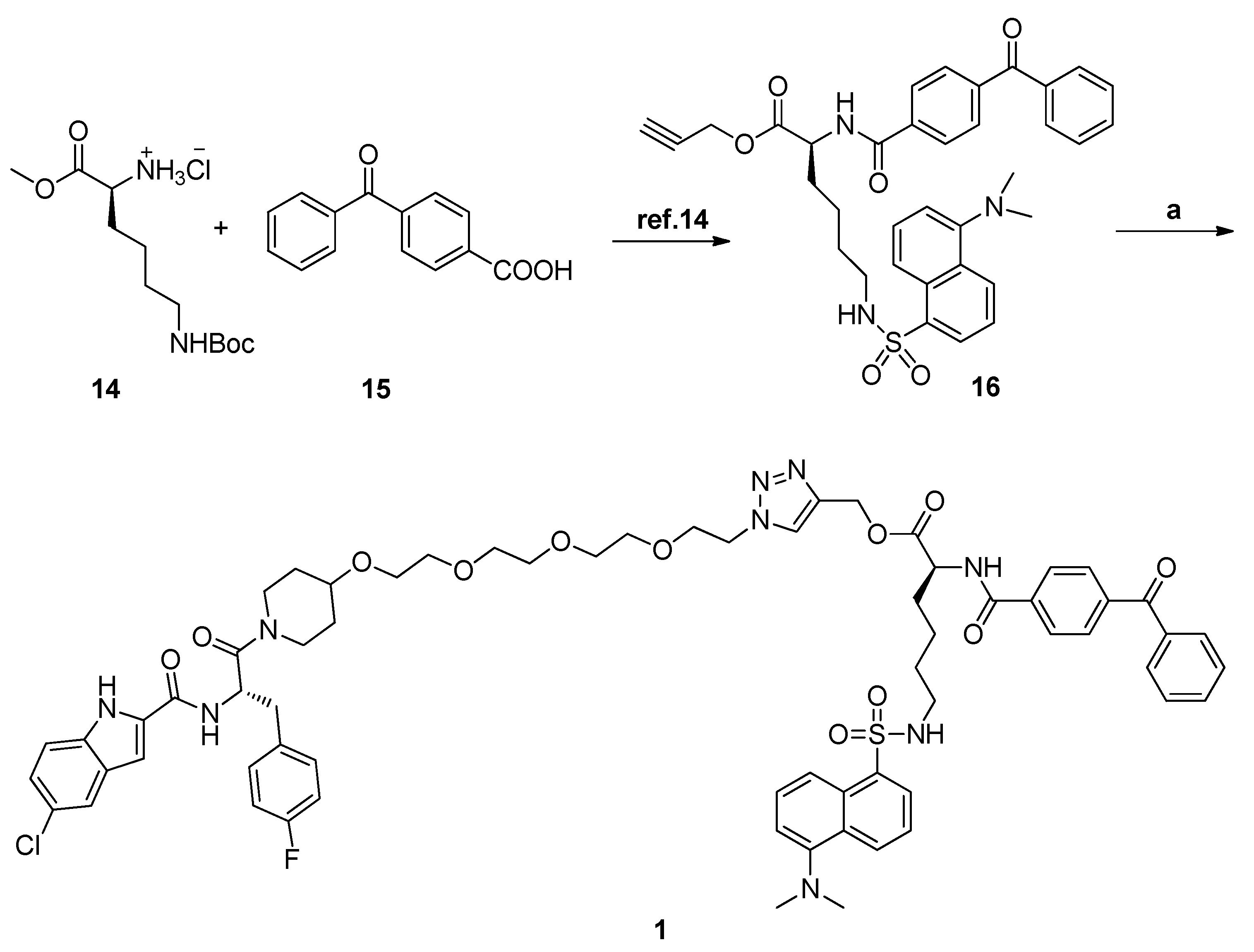
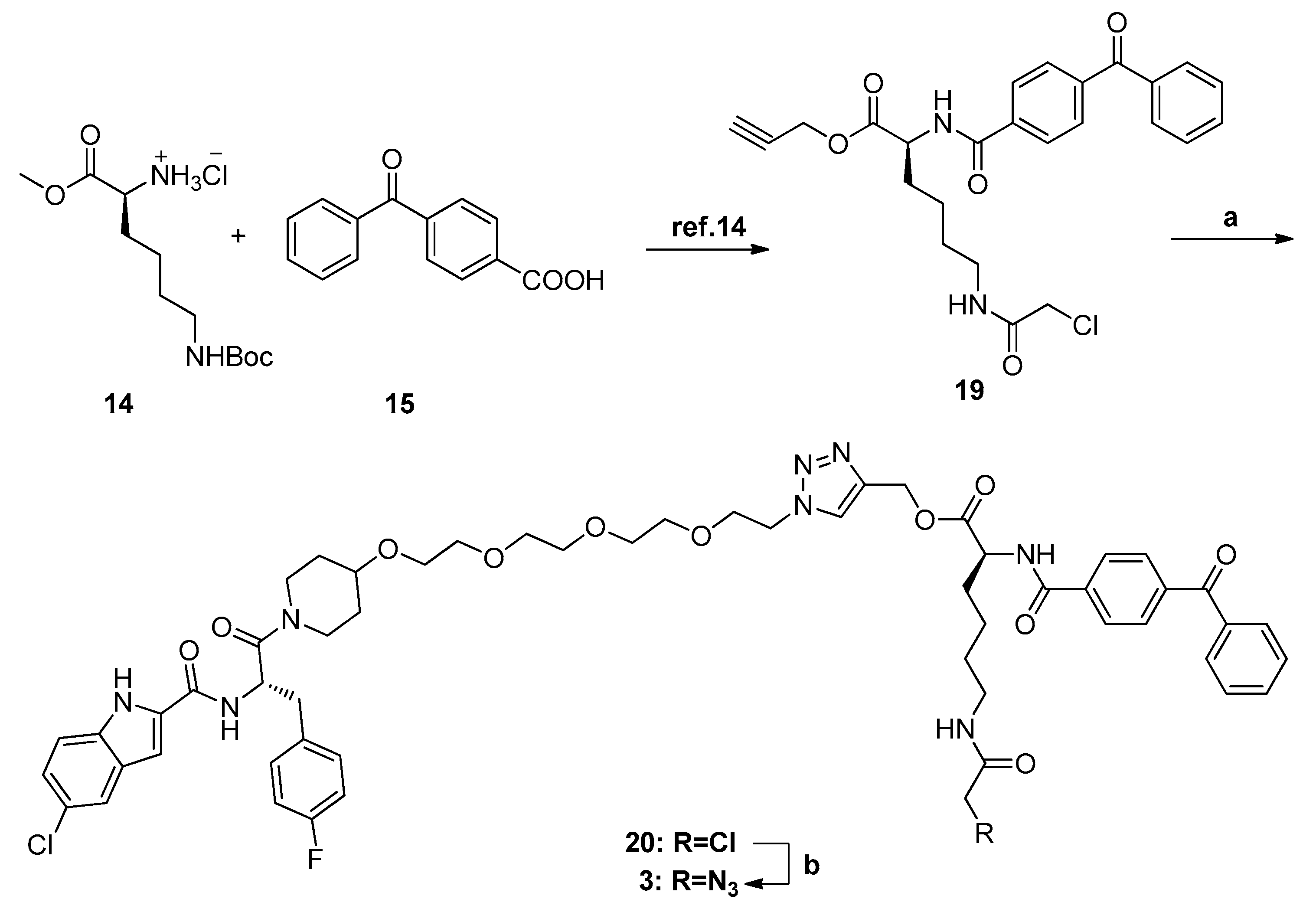
N-[(S)-1-(4-{2-[2-(2-{2-[4-({[(S)-2-(4-Benzoylbenzamido)-6-(2-azidoacetamido)hexanoyl]oxy}methyl)-1H-1,2,3-triazol-1-yl]ethoxy}ethoxy)ethoxy]ethoxy}piperidin-1-yl)-3-(4-fluorophenyl)-1-oxopropan-2-yl]-5-chloro-1H-indole-2-carboxamide (3). HPLC analysis was as follows: 98.8%; m.p. 113–115 °C; 1H-NMR (400 MHz, d6-DMSO) δ 11.74 (d, J = 9.6 Hz, 1H), 8.97–8.91 (m, 2H), 8.13 (d, J = 5.2 Hz, 1H), 8.10 (t, J = 5.6 Hz, 1H), 8.05 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 7.6 Hz, 2H), 7.71 (t, J = 6.8 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.40 (d, J = 8.8 Hz, 1H), 7.34 (t, J = 6.8 Hz, 2H), 7.26 (s, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.06 (t, J = 8.8 Hz, 2H), 5.21 (s, 2H), 5.14–5.07 (m, 1H), 4.58–4.51 (m, 2H), 4.49–4.43 (m, 1H), 4.00–3.97 (m, 1H), 3.83–3.79 (m, 4H), 3.73–3.66 (m, 1H), 3.49–3.45 (m, 13H), 3.31–3.17 (m, 1H), 3.13–2.91 (m, 5H), 1.83–1.65 (m, 4H), and 1.43–1.23 (m, 6H); 13C-NMR (CDCl3, 100 MHz) δ 195.9, 172.3, 169.4, 167.5, 166.5, 161.4 (d, J = 238.3 Hz), 160.6, 142.0, 139.9, 137.5, 137.1, 135.3, 134.5, 133.5, 132.9, 131.7, 131.6, 130.2, 129.9, 129.1, 128.5, 128.2, 125.6, 124.7, 124.0, 121.1, 115.3, 115.1, 114.3, 103.3, 74.6, 73.9, 70.5, 70.2 (d, J = 6.8 Hz), 70.1 (d, J = 7.9 Hz), 70.0 (d, J = 6.4 Hz), 69.1, 67.2 (d, J = 6.9 Hz), 58.3, 53.3, 51.3, 50.6 (d, J = 9.4 Hz), 49.9, 43.1, 42.7, 38.8, 36.9 (d, J = 8.8 Hz), 31.7 (d, J = 34.9 Hz), 31.1 (d, J = 42.8 Hz), 30.5, 29.0, and 23.6. EIMS m/z 1121.0 [M]+.
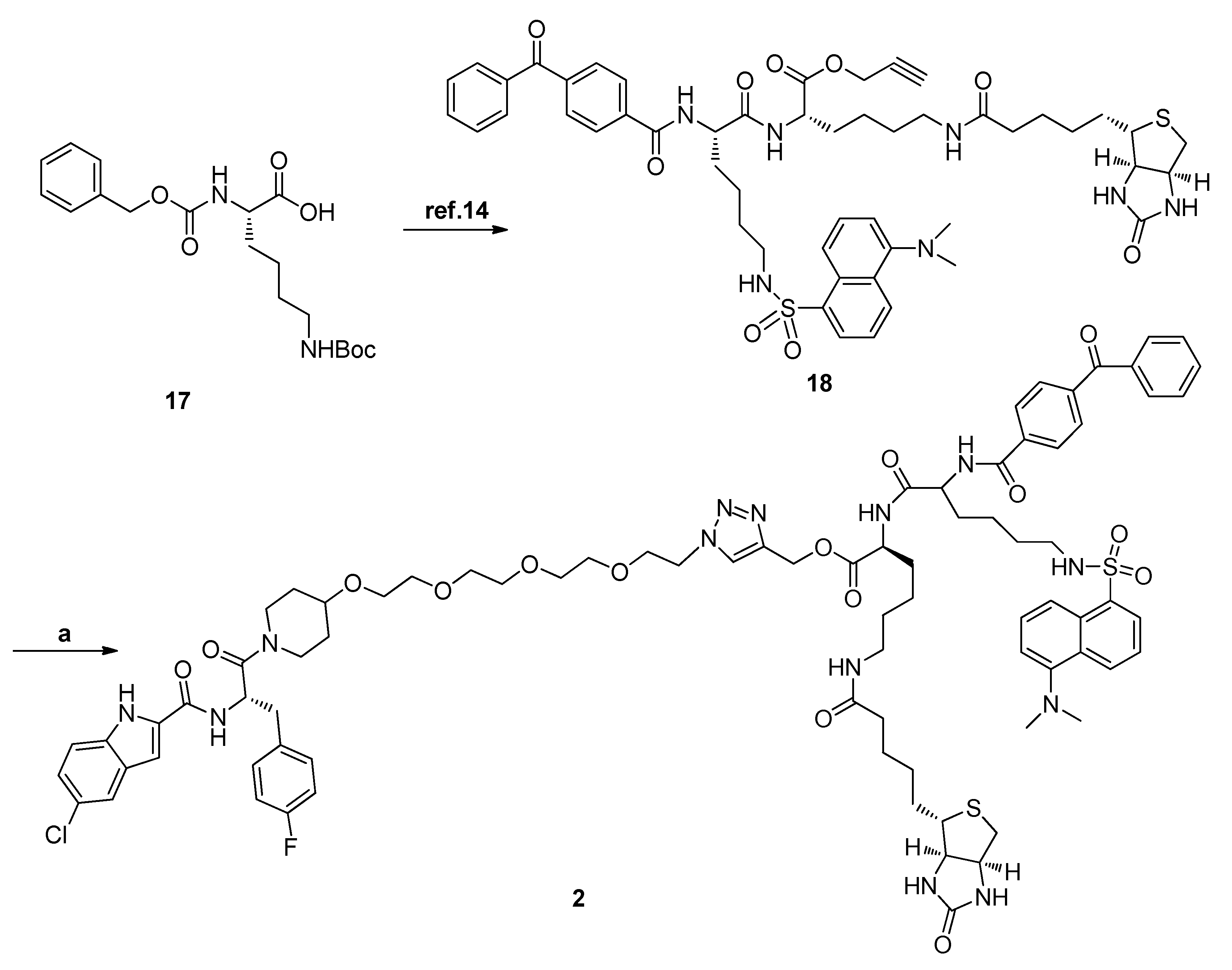
3.1.8. General Procedure for Synthesis of Compounds 1, 2, and 20
CuSO4·5H2O (0.19 equiv.) and Vc-Na (0.4 equiv.) were added to a mixture of compound 13 (1.0 equiv.) and (S)-prop-2-ynyl 2-(4-benzoylbenzamido)-6-(2-chloroacetamido)hexanoate (1.2 equiv.) in CH2Cl2/H2O (4 mL, v:v = 1:1). The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was diluted with EtOAc and washed with water, dried over anhydrous Na2SO4, and evaporated. The residue was purified by HPLC to give the product as a white solid.
N-[(S)-1-(4-{2-[2-(2-{2-[4-({[(S)-2-(4-Benzoylbenzamido)-6-(2-chloro-acetylamino)hexanoyl]oxy}methyl)-1H-1,2,3-triazol-1-yl]ethoxy}ethoxy)ethoxy]ethoxy}piperidin-1-yl)-3-(4-fluorophenyl)-1-oxopropan-2-yl]-5-chloro-1H-indole-2-carboxamide (20). HPLC analysis was as follows: 95.9%; m.p. 137–139 °C; 1H-NMR (400 MHz, d6-DMSO) δ 11.74 (d, J = 9.6 Hz, 1H), 8.97–8.91 (m, 2H), 8.22 (t, J = 5.2 Hz, 1H), 8.13 (d, J = 5.6 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 7.2 Hz, 2H), 7.71 (t, J = 7.6 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.39 (d, J = 8.8 Hz, 1H), 7.34 (t, J = 6.8 Hz, 2H), 7.26 (s, 1H), 7.17 (d, J = 8.8 Hz, 1H), 7.06 (t, J = 8.8 Hz, 2H), 5.20 (s, 2H), 5.15–5.09 (m, 1H), 4.55–4.51 (m, 2H), 4.47–4.42 (m, 1H), 4.02 (s, 2H), 4.00–3.96 (m, 1H), 3.82–3.78 (m, 2H), 3.73–3.65 (m, 1H), 3.49–3.43 (m, 13H), 3.30–3.21 (m, 1H), 3.10–2.93 (m, 5H), 1.84–1.65 (m, 4H), and 1.48–1.23 (m, 6H); 13C-NMR (d6-DMSO, 100 MHz) δ 195.9, 172.3, 169.4, 166.5, 166.2, 161.5 (d, J = 238.3 Hz), 160.6, 142.0, 139.9, 137.5, 137.1, 135.3, 134.5, 133.5, 132.9, 131.7, 131.6, 130.2, 129.9, 129.1, 128.5, 128.2, 125.6, 124.7, 124.0, 121.1, 115.4, 115.1, 114.3, 103.3, 74.6, 73.9, 70.5, 70.2 (d, J = 6.1 Hz), 70.0 (d, J = 3.4 Hz), 69.9 (d, J = 1.7 Hz), 69.1, 67.1 (d, J =7.4 Hz), 58.2, 53.3, 50.7, 49.9, 43.1, 42.7, 39.1, 36.9 (d, J = 8.1 Hz), 31.7 (d, J = 36.1 Hz), 31.1(d, J = 43.4 Hz), 30.4, 28.9, and 23.5. EIMS m/z 1114.0 [M]+.
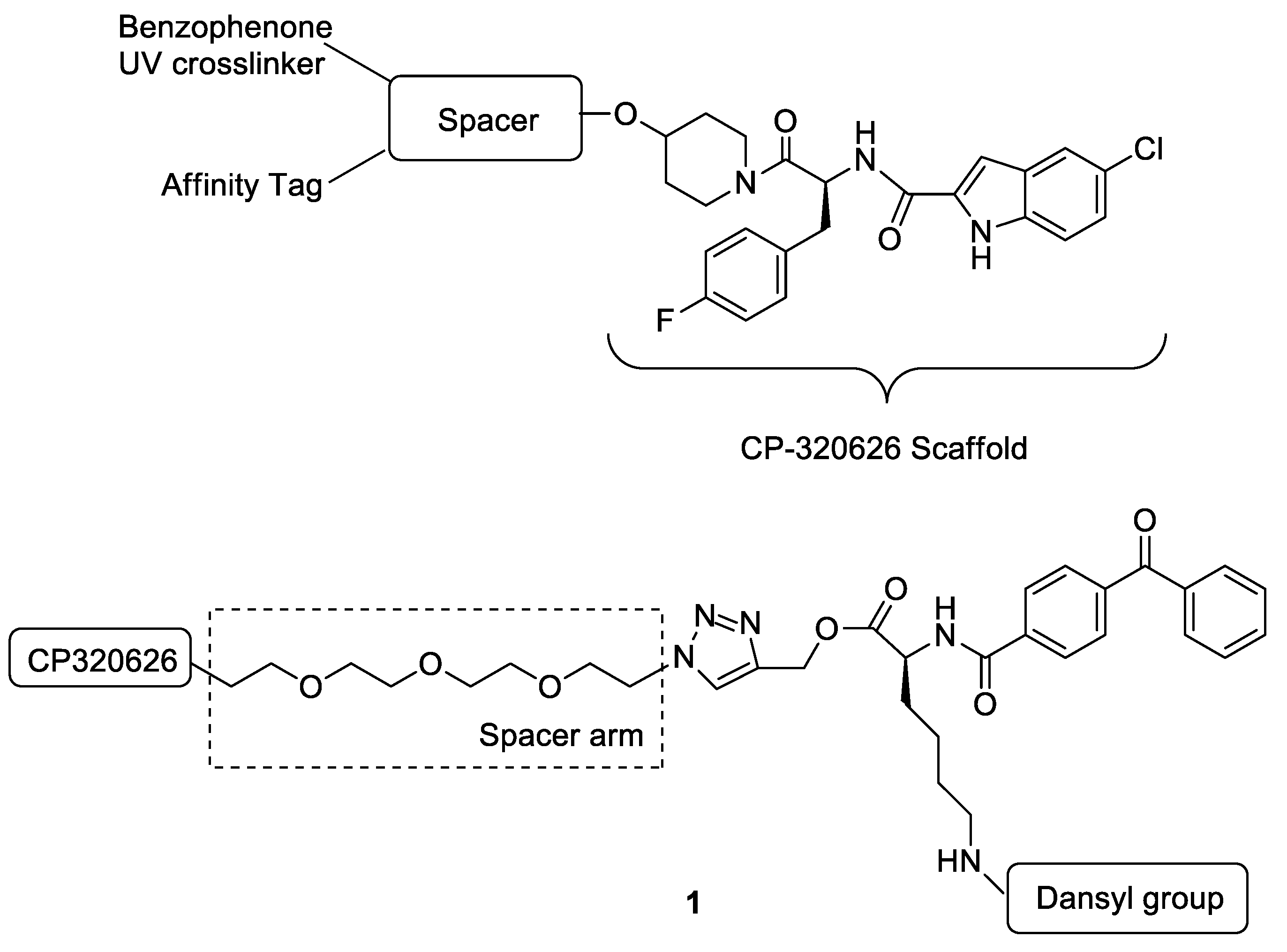
N-[(S)-1-(4-{2-[2-(2-{2-[4-({[(S)-2-(4-Benzoylbenzamido)-6-(dansylamide)hexanoyl]oxy}methyl)-1H-1,2,3-triazol-1-yl]ethoxy}ethoxy)ethoxy]ethoxy}piperidin-1-yl)-3-(4-fluorophenyl)-1-oxopropan-2-yl]-5-chloro-1H-indole-2-carboxamide (1). HPLC analysis was as follows: 100.0%; m.p. 151–153 °C; 1H-NMR (400 MHz, CD3OD) δ 8.73 (d, J = 10.0 Hz, 1H), 8.57 (d, J = 10.8 Hz, 1H), 8.41 (d, J = 11.2 Hz, 1H), 8.23 (d, J = 9.6 Hz, 1H), 8.10 (d, J = 12.0 Hz, 1H), 8.04–8.01 (m, 2H), 7.88–7.82 (m, 4H), 7.71 (t, J = 11.2 Hz, 1H), 7.62–7.55 (m, 5H), 7.44 (d, J = 11.6 Hz, 1H), 7.33–7.20 (m, 4H), 7.13 (s, 1H), 7.02 (t, J = 11.2 Hz, 2H), 5.38–5.33 (m, 3H), 4.81–4.76 (m, 1H), 4.62–4.58 (m, 3H), 3.93–3.81 (m, 4H), 3.63–3.59 (m, 13H), 3.24–3.07 (m, 3H), 2.99–2.92 (m, 8H), 1.90–1.72 (m, 4H), and 1.61–1.40 (m, 6H); 13C-NMR (d6-DMSO, 100 MHz) δ 195.9, 172.2, 169.3, 166.5, 162.7, 160.6, 158.7 (d, J = 30 Hz), 151.8, 142.0, 139.9, 137.4, 137.1, 136.6, 135.3, 133.5, 132.9, 131.7, 131.6, 130.2, 129.9, 129.8, 129.5, 129.4, 129.1, 128.6, 128.5, 128.2, 128.1, 125.6, 124.7, 124.0, 121.1, 119.6, 115.5, 115.3, 115.1, 114.3, 103.4, 74.6, 73.8, 70.4, 70.2 (d, J = 5.8 Hz), 70.1 (d, J = 8.0 Hz), 70.0 (d, J = 5.1 Hz), 69.1, 67.1 (d, J = 7.0 Hz), 58.2, 53.3, 49.9, 49.0, 45.5, 42.6, 30.2, 29.5, 29.2, 27.0, and 23.2. EIMS m/z 1271.0 [M]+.
N-[(S)-1-(4-{2-[2-(2-{2-[4-({[(S)-2-[(S)-2-(4-Benzoylbenzamido)-6-(dansylamide)hexanamido]-6-(5-{(3aR,4S,6aS)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-1-yl}pentanamido)hexanoyl]oxy}methyl)-1H-1,2,3-triazol-1-yl]ethoxy}ethoxy)ethoxy]ethoxy}piperidin-1-yl)-3-(4-fluorophenyl)-1-oxopropan-2-yl]-5-chloro-1H-indole-2-carboxamide (2). HPLC analysis was as follows: 99.4%; m.p. 159–161 °C; 1H-NMR (400 MHz, d6-DMSO) δ 11.74 (d, J = 10.0 Hz, 1H), 8.98–8.92 (m, 1H), 8.58 (d, J = 7.6 Hz, 1H), 8.44 (d, J = 8.8 Hz, 1H), 8.34 (d, J = 7.2 Hz, 1H), 8.30 (d, J = 8.8 Hz, 1H), 8.11–8.09 (m, 2H), 8.02 (d, J = 8.0 Hz, 2H), 7.91 (t, J = 5.6 Hz, 1H), 7.81–7.69 (m, 7H), 7.63–7.54 (m, 4H), 7.39 (dd, J = 8.8, 1.6 Hz, 1H), 7.34 (t, J = 6.4 Hz, 2H), 7.26 (s, 1H), 7.23 (d, J = 7.6 Hz, 1H), 7.17 (d, J = 8.8 Hz, 1H), 7.06 (t, J = 8.8 Hz, 2H), 6.43 (br s, 1H), 6.37 (br s, 1H), 5.18–5.11 (m, 3H), 4.53–4.49 (m, 2H), 4.45–4.40 (m, 1H), 4.28 (t, J = 6.4 Hz, 1H), 4.24–4.21 (m, 1H), 4.11–4.08 (m, 1H), 4.00–3.97 (m, 1H), 3.82–3.77 (m, 2H), 3.72–3.64 (m, 1H), 3.50–3.44 (m, 13H), 3.30–3.18 (m, 1H), 3.13–2.90 (m, 6H), 2.81 (s, 6H), 2.78–2.76 (m, 3H), 2.56 (d, J = 12.4 Hz, 1H), 2.03 (t, J = 7.2 Hz, 2H), 1.73–1.56 (m, 6H), and 1.48–1.24 (m, 16H); 13C-NMR (d6-DMSO, 100 MHz) δ 195.9, 172.5, 172.3, 172.2, 169.4, 166.2, 163.2, 161.5 (d, J = 241.1 Hz), 160.6, 151.8, 141.9, 139.7, 137.9, 137.2, 136.6, 135.3, 135.1, 134.4, 133.5, 132.9, 131.7, 131.6, 130.2, 129.8, 129.6, 129.5, 129.1, 128.6, 128.5, 128.2, 128.1, 125.5, 124.7, 124.0, 121.1, 119.6, 115.5, 115.3, 115.1, 114.3, 103.4, 74.6, 73.9, 70.5, 70.2 (d, J = 6.2 Hz), 70.1 (d, J = 3.4 Hz), 70.0 (d, J = 1.7 Hz), 69.1, 67.1 (d, J = 7.1 Hz), 61.5, 59.7, 58.1, 55.9, 53.6, 52.4, 50.5, 49.9, 45.5, 43.1, 42.9, 38.6, 35.7, 31.9, 31.4 (d, J = 6.2 Hz), 30.9 (d, J = 10.9 Hz), 29.6, 29.2, 28.7, 28.5,25.8, 23.3, and 23.2. EIMS m/z 1625.0 [M]+.

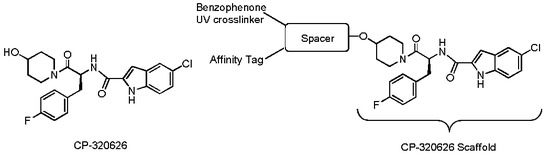











{kind=link}
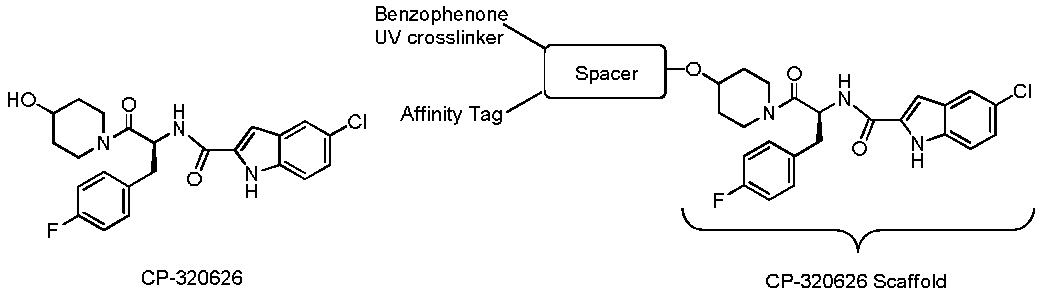
{kind=link}
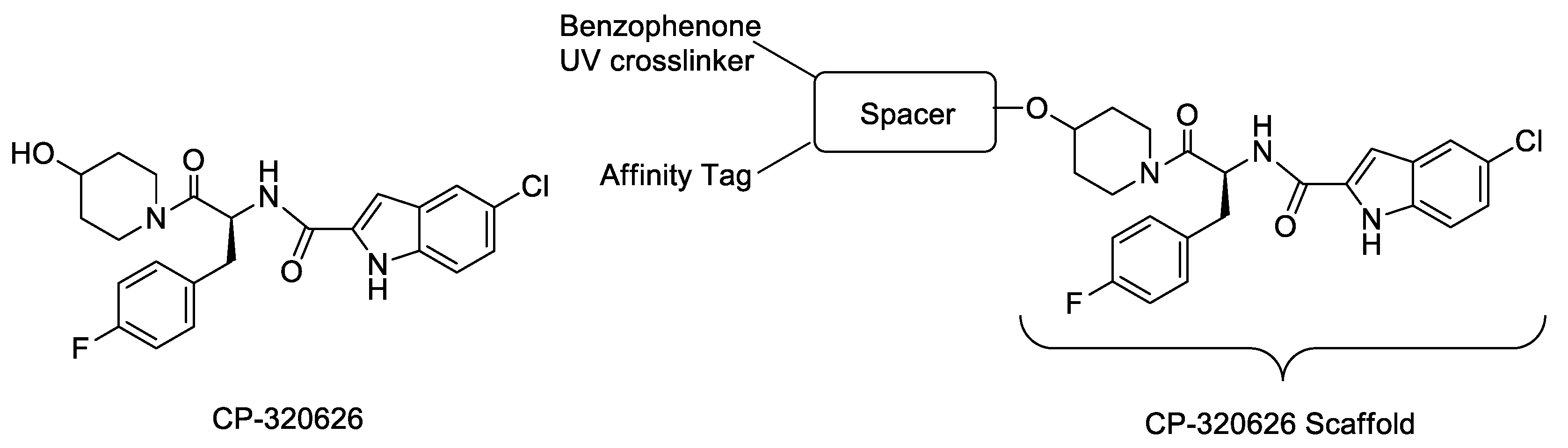
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}