
Isomers of β,β-Dinitro-5,10,15,20-tetraphenylporphyrin Derivatives: Valuable Starting Materials for Further Transformations


Abstract
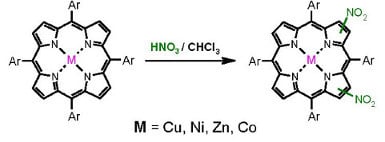
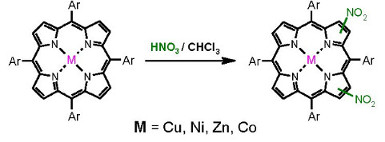
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
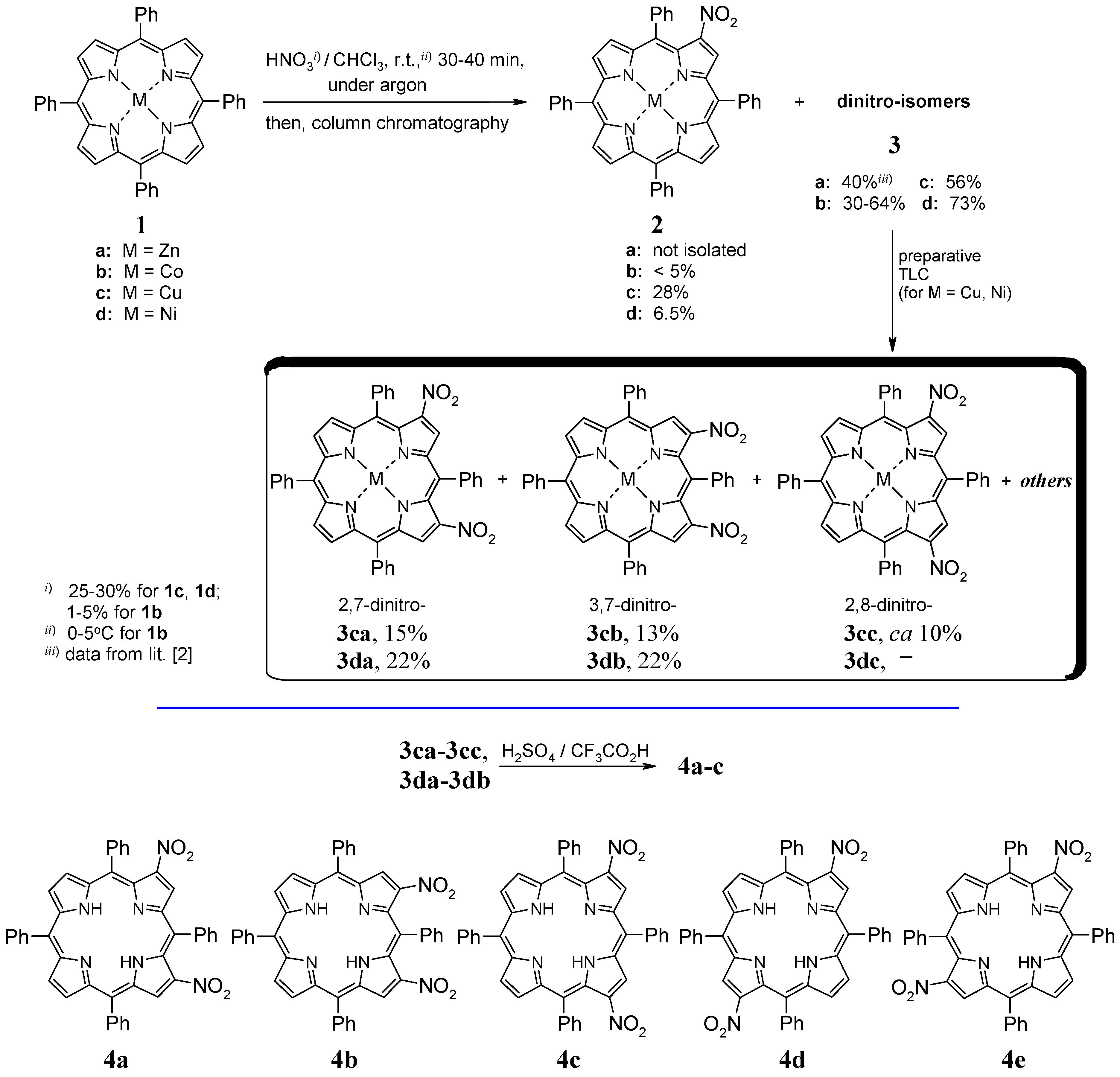
2.1. Nitration of Cobalt(II) and Copper(II) Complexes
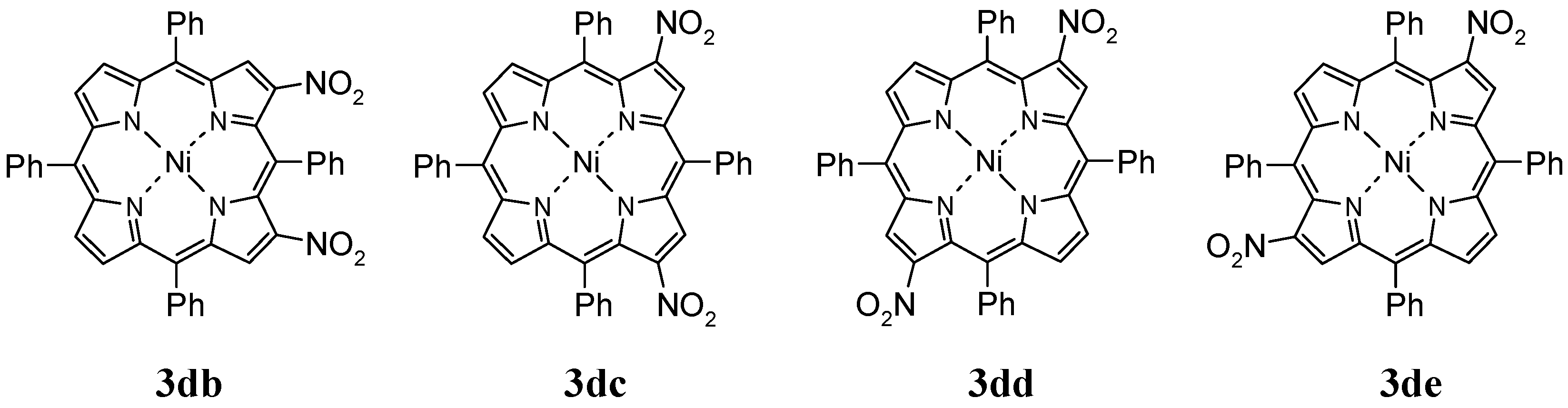
2.2. Nitration of Nickel(II) Complex
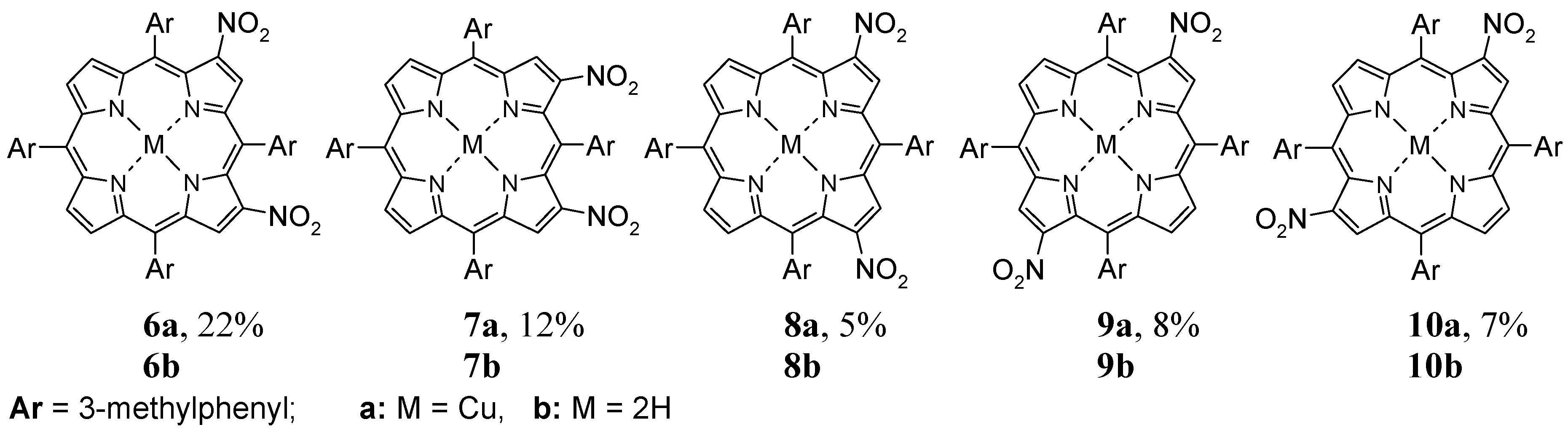
2.3. Nitration of 5,10,15,20-tetrakis(3-Methylphenyl)porphyrin–Copper(II) Complex (5)
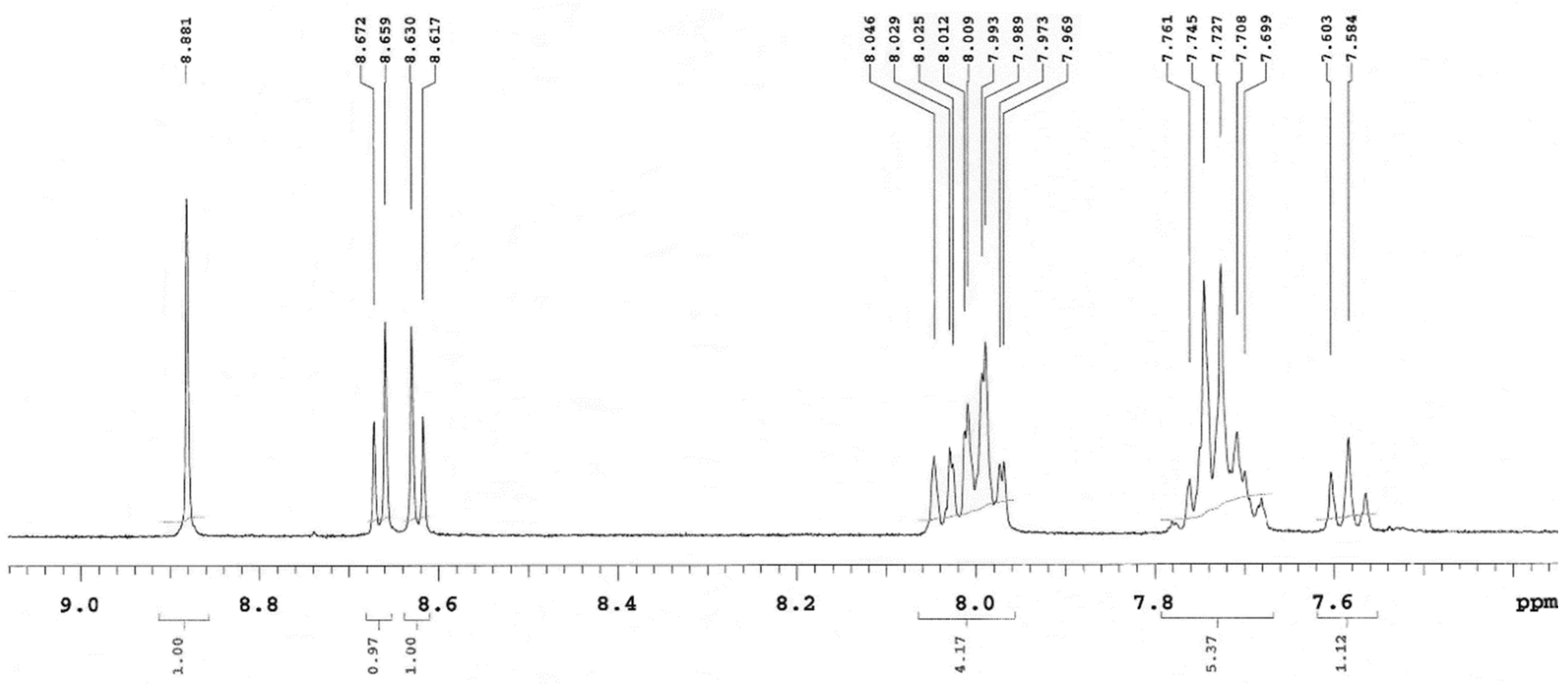
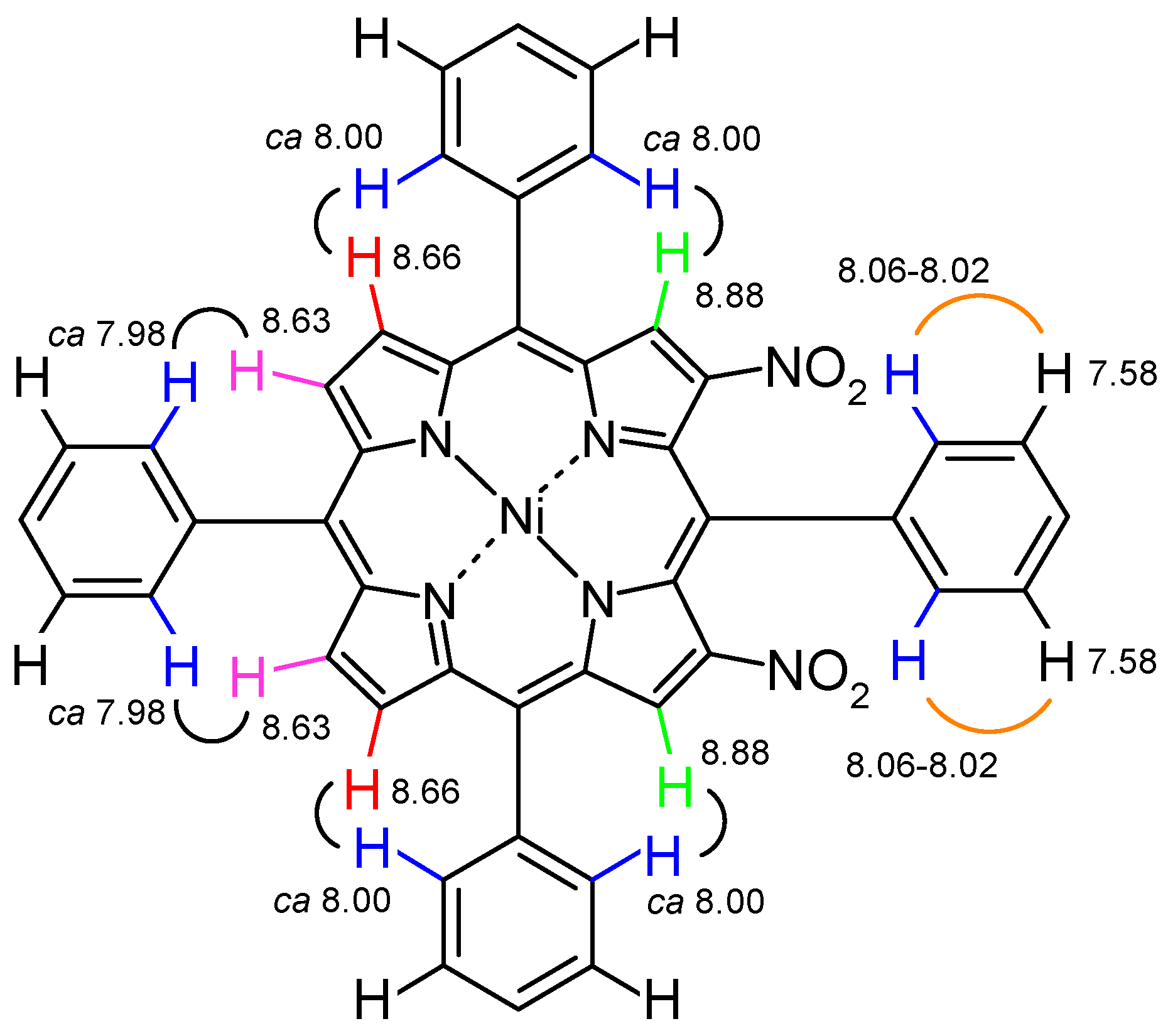
2.4. 1H NMR Spectra and Structure Elucidation of the Isomers
3. Experimental
3.1. Materials and Methods—General.
3.2. Procedures and Data of New Compounds
3.2.1. Nitration of Copper(II) Complex of 5,10,15,20-Tetraphenylporphyrin (1c)
Products Data
3.2.2. Nitration of Nickel(II) Complex of 5,10,15,20-Tetraphenylporphyrin (1d)
Products Data
3.2.3. Demetallation
Products Data
3.2.4. Nitration of Copper(II) Complex of 5,10,15,20-tetrakis(3-Methylphenyl)porphyrin (5)
Products Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Handbook of Porphyrin Science; Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) World Scientific Publishing Co.: New Jersey, NJ, USA; London, UK; Singapore; Beijing, China; Shanghai, China; Hong Kong; Taipei, Taiwan; Chennai, India, 2010–2012; Volume 1–25.
- Ostrowski, S. Preparation of 2-nitro-5,10,15,20-tetraphenylporphyrin zinc(II) complex—post scriptum to electrophilic nitration of porphyrins at the β-pyrrolic position. Polish J. Chem. 2005, 79, 1169–1172. [Google Scholar]
- Mikus, A.; Ostrowski, S. Synthesis of highly functionalized porphyrins—substituted in all β-positions of EASTERN HALF. In Proceedings of the Petra International Conference of Chemistry (PICC) and Transmediterranean Colloquium on Heterocyclic Chemistry (TRAMECH–5), Tafila, Jordan, 25–28 June 2007; p. 28. [Google Scholar]
- Ostrowski, S.; Wyrębek, P. The first example of Diels-Alder cycloaddition of ortho-xylylenes to meso-tetraarylporphyrins containing electron-deficient β,β-double bonds. Tetrahedron Lett. 2006, 47, 8437–8440. [Google Scholar] [CrossRef]
- Fang, Y.; Jiang, X.; Ou, Z.; Michelin, C.; Desbois, N.; Gros, C.P.; Kadish, K.M. Redox properties of nitro-phenylporphyrins and electrosynthesis of nitrophenyl-linked Zn porphyrin dimers or arrays. J. Porphyrins Phthalocyanines 2014, 18, 832–841. [Google Scholar] [CrossRef]
- Dahal, S.; Krishnan, V.; Nethaji, M. Isomers of β-substituted di and tri-nitrotetraphenylporphyrins and their copper(II) derivatives: Structure, optical and electrochemical redox properties. Proc. Indian Acad. Sci. Chem. Sci. 1998, 110, 37–52. [Google Scholar] [CrossRef]
- Photodynamic Tumor Therapy: 2nd and 3rd Generation Photosensitizers; Moser, J.G. (Ed.) Harwood Academic Publ.: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Vaz Serra, V.I.; Pires, S.M.G.; Alonso, C.M.A.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S. meso-Tetraarylporphyrins bearing nitro or amino groups: Synthetic strategies and reactivity profiles. Top. Heterocycl. Chem. 2014, 33, 35–78. [Google Scholar]
- Taniguchi, M.; Lindsey, J.S. Synthetic chlorins, possible surrogates for chlorophylls, prepared by derivatization of porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Wyrębek, P.; Osuch-Kwiatkowska, A.; Pakulski, Z.; Jarosz, S.; Ostrowski, S. Synthesis of sugar-decorated hydrophilic porphyrins. J. Porphyrins Phthalocyanines 2013, 17, 384–391. [Google Scholar] [CrossRef]
- Pandey, R.K. Recent advances in photodynamic therapy. J. Porphyrins Phthalocyanines 2000, 4, 368–373. [Google Scholar] [CrossRef]
- Ostrowski, S.; Szerszeń, D.; Ryszczuk, M. Electrophilic nitration of meso-tetraarylporphyrin complexes at the β-pyrrolic position. Synthesis 2005, 37, 819–823. [Google Scholar] [CrossRef]
- Wyrȩbek, P.; Ostrowski, S. Synthesis of some β-nitro-meso-tetraphenylporphyrin derivatives. J. Porphyrins Phthalocyanines 2007, 11, 822–828. [Google Scholar] [CrossRef]
- Mikus, A.; Zając, M.; Ostrowski, S. Frontiers in the electrophilic nitration of meso-tetraphenylporphyrin derivatives. Org. Chem. Front. 2018, 5, 2840–2844. [Google Scholar] [CrossRef]
- Giraudeau, A.; Callot, H.J.; Jordan, J.; Ezhar, I.; Gross, M. Substituent effects in the electroreduction of porphyrins and metalloporphyrins. J. Am. Chem. Soc. 1979, 101, 3857–3862. [Google Scholar] [CrossRef]
- Hombrecher, H.K.; Gherdan, V.M.; Ohm, S.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S.; Condesso, M.F. Synthesis and electrochemical investigation of β-alkyloxy substituted meso-tetraphenylporphyrins. Tetrahedron 1993, 49, 8569–8578. [Google Scholar] [CrossRef]
- Alonso, C.A.A.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S.; Hombrecher, H.K. Reaction of β-amino-meso-tetraphenylporphyrin with α,β-unsaturated carbonyl compounds: An approach to fused pyridino-porphyrins. Tetrahedron Lett. 1997, 38, 2757–2758. [Google Scholar] [CrossRef]
- Evans, B.; Smith, K.M.; Cavaleiro, J.A.S. Bile pigment studies. Part 4. Some novel reactions of metallo-porphyrins with thallium(III) and cerium(IV) salts. Ring cleavage of meso-tetraphenylporphyrin. J. Chem. Soc. Perkin Trans. 1 1978, 768–773. [Google Scholar] [CrossRef]
- Catalano, M.M.; Crossley, M.J.; Harding, M.M.; King, L.G. Control of reactivity at the porphyrin periphery by metal ion co-ordination: A general method for specific nitration at the β-pyrrolic position of 5,10,15,20-tetraaryl-porphyrins. J. Chem. Soc. Chem. Commun. 1984, 22, 1535–1536. [Google Scholar] [CrossRef]
- Shea, K.M.; Jaquinod, L.; Smith, K.M. Dihydroporphyrin synthesis: New methodology. J. Org. Chem. 1998, 63, 7013–7021. [Google Scholar] [CrossRef] [PubMed]
- Wyrębek, P. Modification of porphyrin ring with the use of cycloaddition reactions. PhD Thesis, Uniwersytet Przyrodniczo-Humanistyczny, Siedlce, Poland, 2011. [Google Scholar]
- Smith, K.M.; Bisset, G.M.F.; Bushell, M.J. Partial syntheses of optically pure methyl bacteriopheophorbides c and d from methyl pheophorbide a. J. Org. Chem. 1980, 45, 2218–2224. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikus, A.; Rosa, M.; Ostrowski, S. Isomers of β,β-Dinitro-5,10,15,20-tetraphenylporphyrin Derivatives: Valuable Starting Materials for Further Transformations. Molecules 2019, 24, 838. https://doi.org/10.3390/molecules24050838
Mikus A, Rosa M, Ostrowski S. Isomers of β,β-Dinitro-5,10,15,20-tetraphenylporphyrin Derivatives: Valuable Starting Materials for Further Transformations. Molecules. 2019; 24(5):838. https://doi.org/10.3390/molecules24050838
Chicago/Turabian StyleMikus, Agnieszka, Mariusz Rosa, and Stanisław Ostrowski. 2019. "Isomers of β,β-Dinitro-5,10,15,20-tetraphenylporphyrin Derivatives: Valuable Starting Materials for Further Transformations" Molecules 24, no. 5: 838. https://doi.org/10.3390/molecules24050838
APA StyleMikus, A., Rosa, M., & Ostrowski, S. (2019). Isomers of β,β-Dinitro-5,10,15,20-tetraphenylporphyrin Derivatives: Valuable Starting Materials for Further Transformations. Molecules, 24(5), 838. https://doi.org/10.3390/molecules24050838