
Differentiation between Enamines and Tautomerizable Imines Oxidation Reaction Mechanism using Electron-Vibration-Vibration Two Dimensional Infrared Spectroscopy


Abstract
:1. Introduction
2. Results
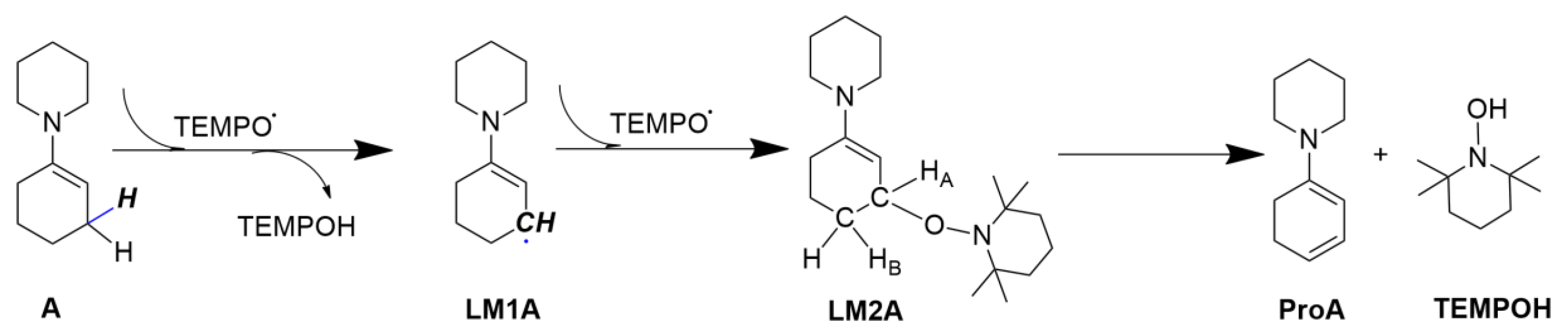
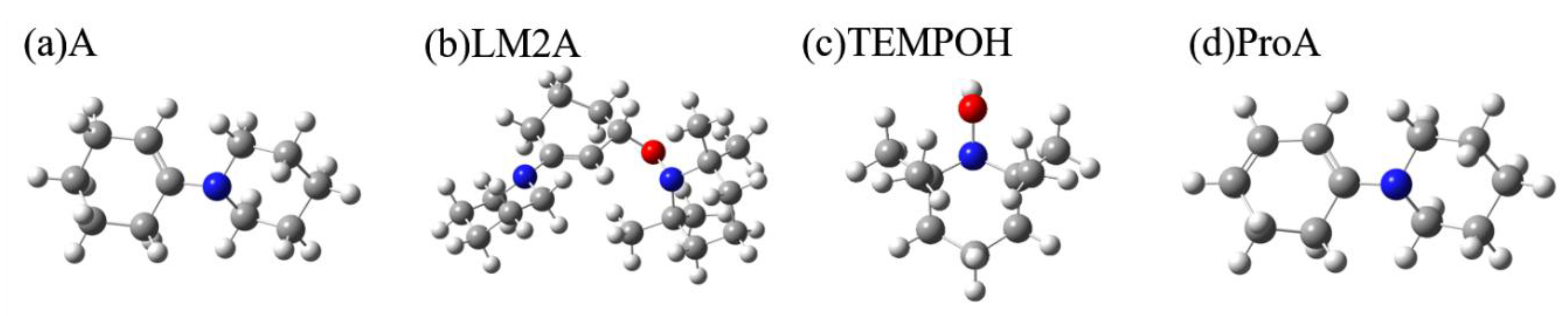
2.1. Calculating Vibrational Spectra of Key Species along the Pathway of Enamine Oxidation Reaction
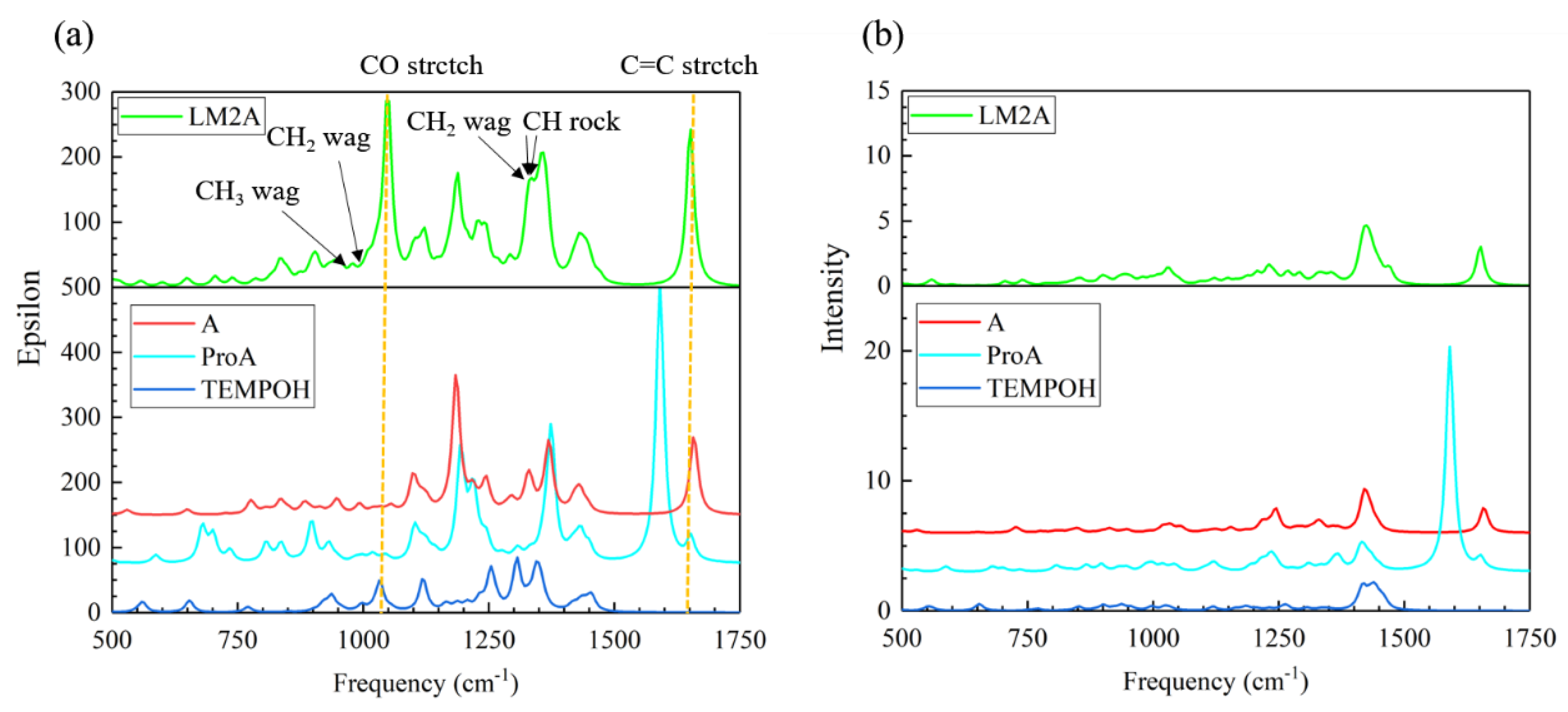
2.1.1. Calculated IR and Raman Spectra
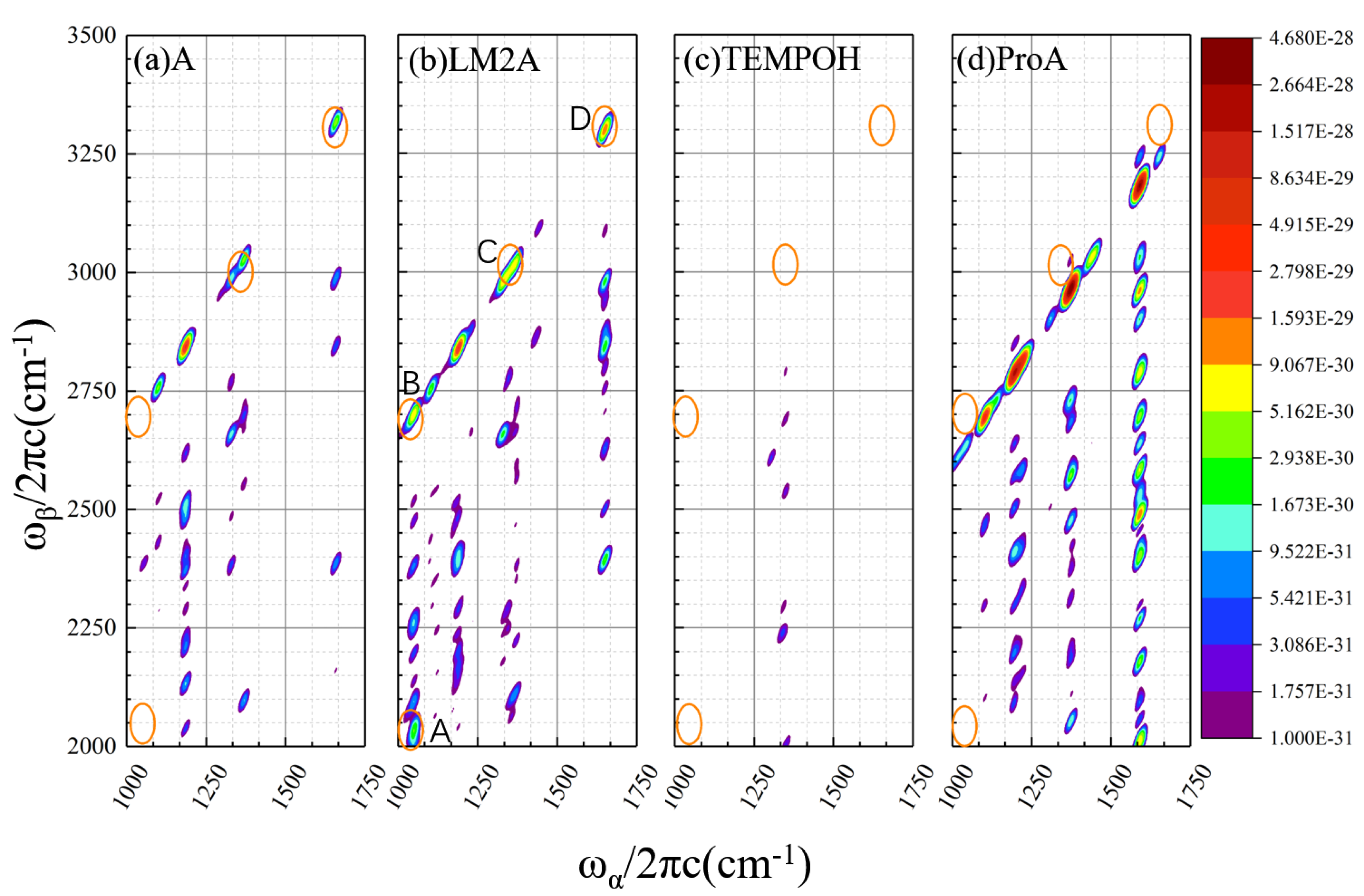
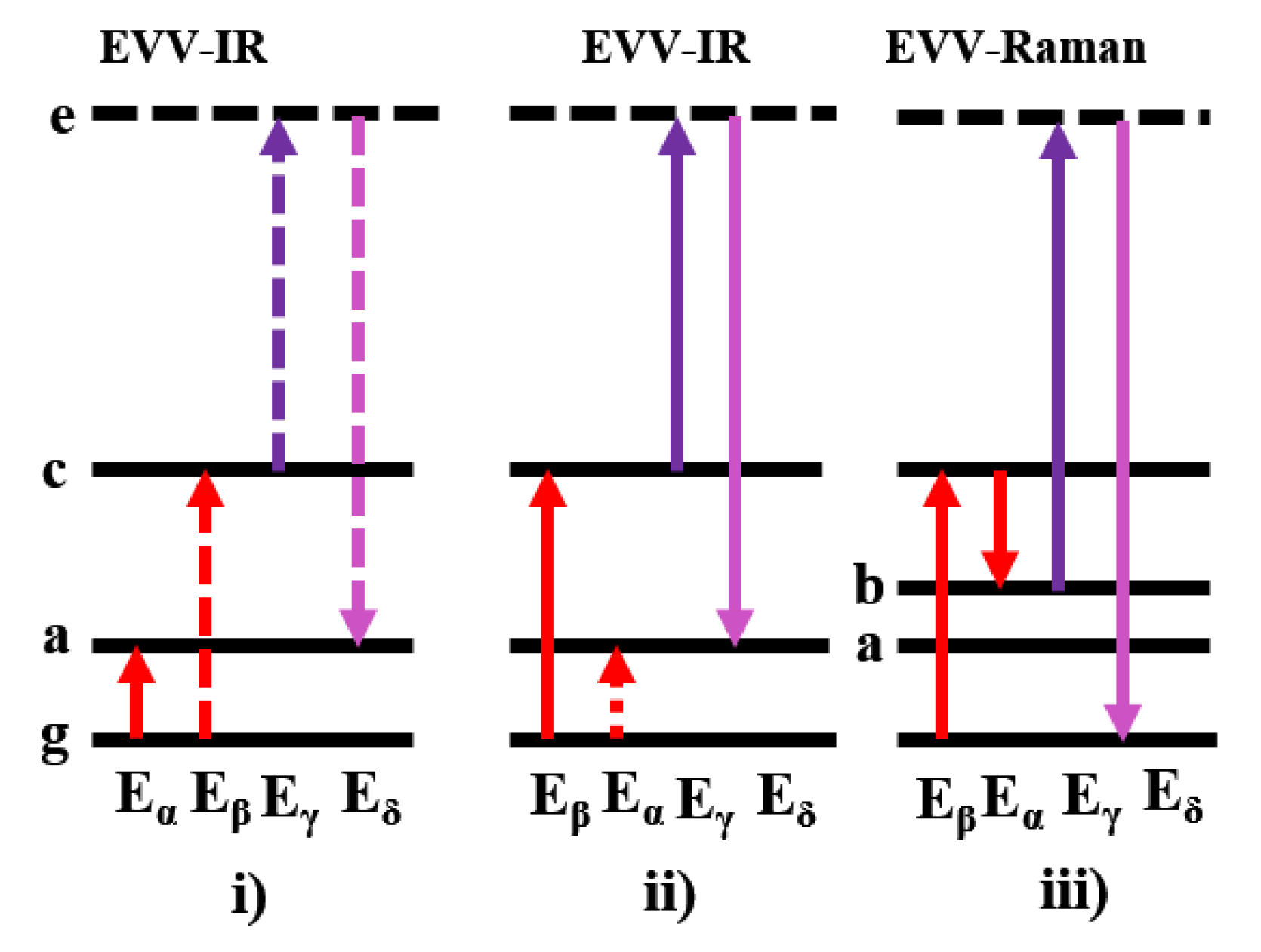
2.1.2. Calculated EVV-2DIR Spectrum in Enamine Reaction Pathway
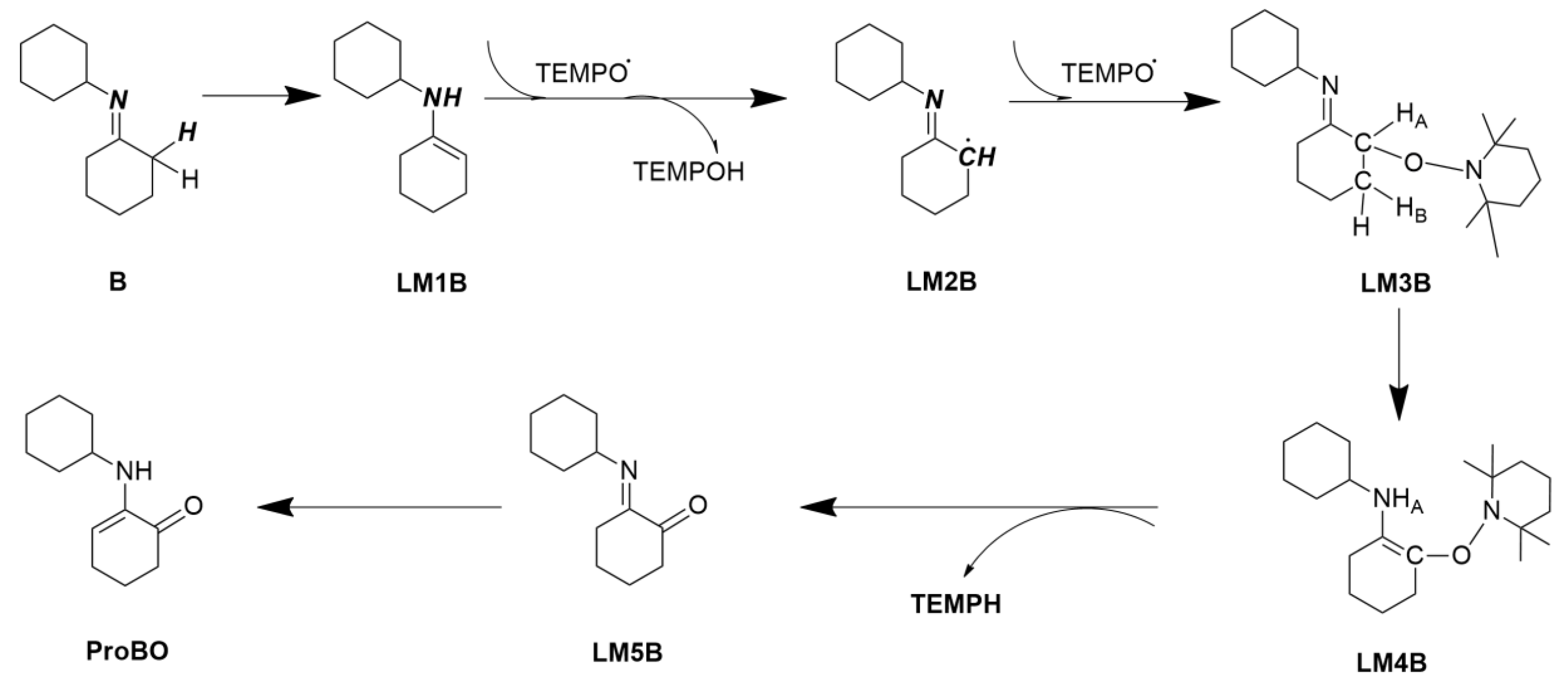
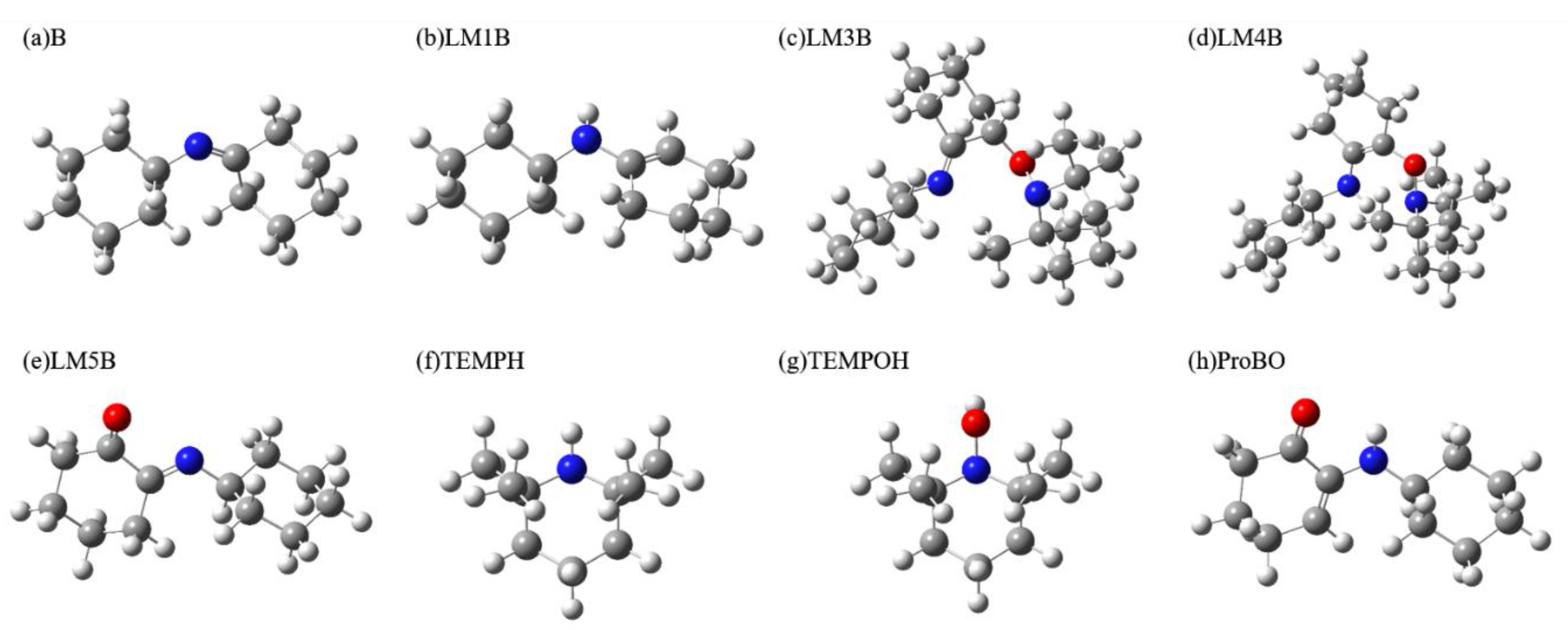
2.2. Calculating Vibrational Spectra of Key Species along the Pathway of Imine Oxidation Reaction
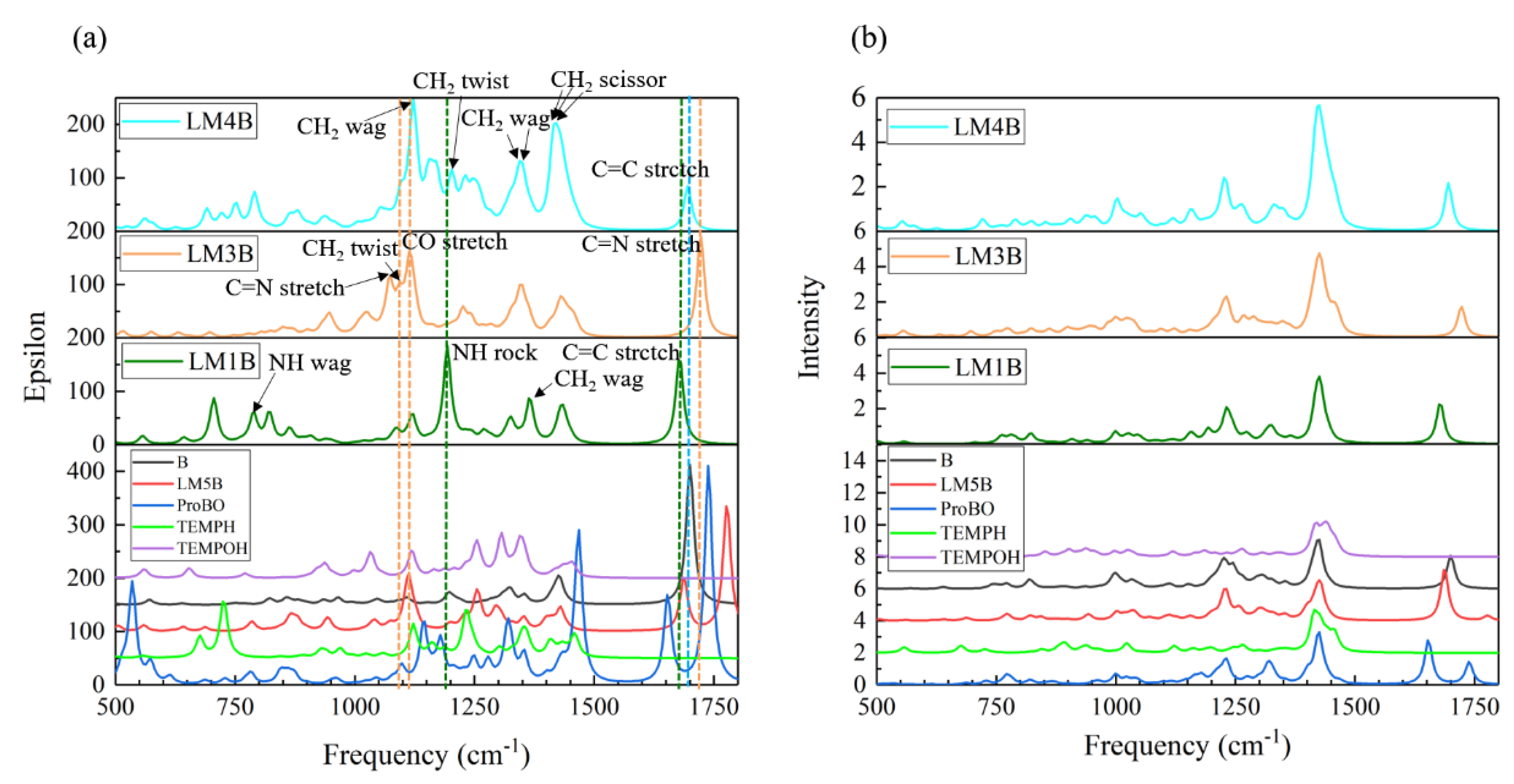
2.2.1. Calculated IR and Raman Spectrum in the Imine Reaction Pathway
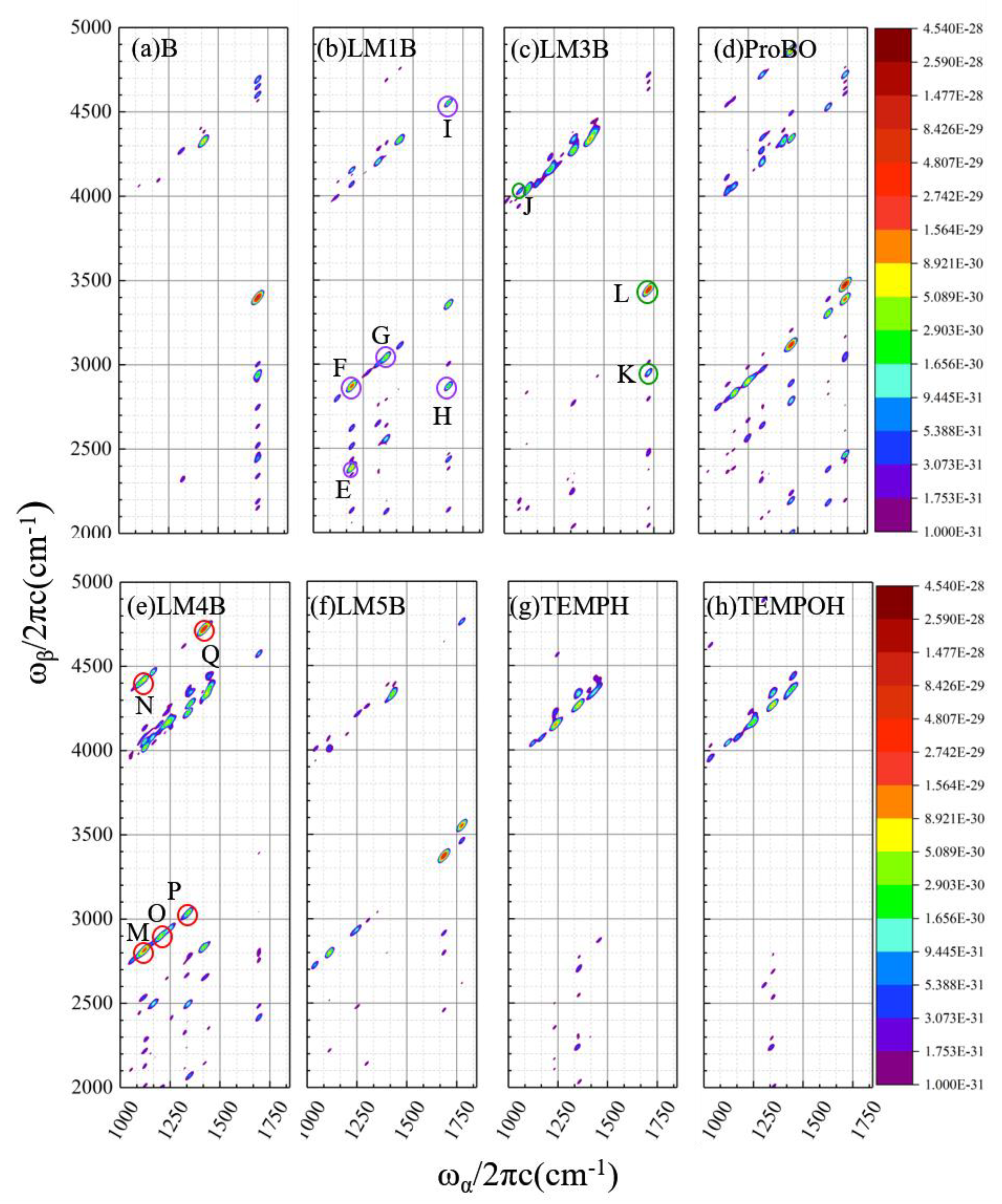
2.2.2. Calculated EVV-2DIR Spectrum in Imine Reaction Pathway
2.2.3. Analysis of the Characteristic Cross Peaks of Intermediates in Oxidation Reaction of Imine
3. Discussion
4. Materials and Methods
4.1. EVV-2DIR Spectroscopy
4.2. DFT calculations and EVV-2DIR Spectroscopy Theoretical Simulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malinauskas, A.; Holze, R. UV-VIS spectroelectrochemical detection of intermediate species in the electropolymerization of an aniline derivative. Electrochim. Acta 1998, 43, 2413–2422. [Google Scholar] [CrossRef]
- Tang, F.; Zhang, Y.; Rath, N.P.; Mirica, L.M. Detection of Pd(III) and Pd(IV) Intermediates during the Aerobic Oxidative C-C Bond Formation from a Pd(II) Dimethyl Complex. Organometallics 2012, 31, 6690–6696. [Google Scholar] [CrossRef]
- Fan, J.F.; Yates, J.T. Mechanism of photooxidation of trichloroethylene on TiO2: Detection of intermediates by infrared spectroscopy. J. Am. Chem. Soc. 1996, 118, 4686–4692. [Google Scholar] [CrossRef]
- Platero, E.E.; Mentruit, M.P. Acetylene polymerization on sulfated zirconia: Detection of intermediates by infrared spectroscopy. Catal. Lett. 1997, 45, 59–63. [Google Scholar] [CrossRef]
- Cao, S.; Zhou, X.; Yang, Y.; Zhong, W.; Sun, T. Selective Substitution of 31/42-OH in Rapamycin Guided by an in Situ IR Technique. Molecules 2014, 19, 7770–7784. [Google Scholar] [CrossRef] [PubMed]
- Banert, K.; Hagedorn, M.; Wu, Z.; Zeng, X. Synthesis, Characterization and Reactions of (Azidoethynyl)trimethylsilane. Molecules 2015, 20, 21328–21335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, V.P.; Pandit, S.; Green, A.M.; Trongsiriwat, N.; Wash, P.J.; Klippenstein, S.J.; Lester, M.I. Four-Carbon Criegee Intermediate from Isoprene Ozonolysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production. J. Am. Chem. Soc. 2018, 140, 10866–10880. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, S.L.; Klein, M.P. Detection of a Paramagnetic Intermediate in the S1-State of the Photosynthetic Oxygen-Evolving Complex. J. Am. Chem. Soc. 1992, 114, 2821–2826. [Google Scholar] [CrossRef]
- Gunderson, W.A.; Zatsman, A.I.; Emerson, J.P.; Farquhar, E.R.; Que, L., Jr.; Lipscomb, J.D.; Hendrich, M.P. Electron paramagnetic resonance detection of intermediates in the enzymatic cycle of an extradiol dioxygenase. J. Am. Chem. Soc. 2008, 130, 14465–14467. [Google Scholar] [CrossRef] [PubMed]
- Barbierikova, Z.; Dvoranova, D.; Bella, M.; Milata, V.; Czimerova, A.; Brezova, V. Fused-Ring Derivatives of Quinoxalines: Spectroscopic Characterization and Photoinduced Processes Investigated by EPR Spin Trapping Technique. Molecules 2014, 19, 12078–12098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzen, S.; Gurer-Orhan, H.; Saso, L. Detection of Reactive Oxygen and Nitrogen Species by Electron Paramagnetic Resonance (EPR) Technique. Molecules 2017, 22, 181. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Ma, W.H.; Lei, P.X.; Zhao, J.C. Detection of intermediates in the TiO2-assisted photodegradation of Rhodamine B under visible light irradiation. J. Environ. Sci. 2007, 19, 892–896. [Google Scholar] [CrossRef]
- Marinescu, S.C.; Winkler, J.R.; Gray, H.B. Molecular mechanisms of cobalt-catalyzed hydrogen evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15127–15131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, O.; Hamann, T.W. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 2016, 8, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-B.; Schuman, D.P.; Yang, Y.-F.; Toutov, A.A.; Liang, Y.; Klare, H.F.T.; Nesnas, N.; Oestreich, M.; Blackmond, D.G.; Virgil, S.C.; et al. Potassium tert-Butoxide-Catalyzed Dehydrogenative C-H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study. J. Am. Chem. Soc. 2017, 139, 6867–6879. [Google Scholar] [CrossRef] [PubMed]
- Cowan, M.L.; Bruner, B.D.; Huse, N.; Dwyer, J.R.; Chugh, B.; Nibbering, E.T.; Elsaesser, T.; Miller, R.J. Ultrafast memory loss and energy redistribution in the hydrogen bond network of liquid H2O. Nature 2005, 434, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Loparo, J.J.; Roberts, S.T.; Tokmakoff, A. Multidimensional infrared spectroscopy of water. I. Vibrational dynamics in two-dimensional IR line shapes. J. Chem. Phys. 2006, 125, 194521. [Google Scholar] [CrossRef] [PubMed]
- Loparo, J.J.; Roberts, S.T.; Tokmakoff, A. Multidimensional infrared spectroscopy of water. II. Hydrogen bond switching dynamics. J. Chem. Phys. 2006, 125, 194522. [Google Scholar] [CrossRef] [PubMed]
- Woutersen, S.; Mu, Y.G.; Stock, G.; Hamm, P. Subpicosecond conformational dynamics of small peptides probed by two-dimensional vibrational spectroscopy. Proc. Natl. Acad. Sci. USA 2001, 98, 11254–11258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Kwak, K.; Xie, J.; Fayer, M.D. Ultrafast carbon-carbon single-bond rotational isomerization in room-temperature solution. Science 2006, 313, 1951–1955. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hochstrasser, R.M. Chemical exchange 2D IR of hydrogen-bond making and breaking. Proc. Natl. Acad. Sci. USA 2005, 102, 11185–11190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Kwak, K.; Asbury, J.; Chen, X.; Piletic, I.R.; Fayer, M.D. Ultrafast dynamics of solute-solvent complexation observed at thermal equilibrium in real time. Science 2005, 309, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.R.; Kwak, K.; Chen, X.; Asbury, J.B.; Fayer, M.D. Formation and dissociation of intra-intermolecular hydrogen-bonded solute-solvent complexes: Chemical exchange two-dimensional infrared vibrational echo spectroscopy. J. Am. Chem. Soc. 2006, 128, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Hayashi, T.; Mukamel, S. Coherent Multidimensional Vibrational Spectroscopy of Biomolecules: Concepts, Simulations, and Challenges. Angew. Chem. Int. Ed. 2009, 48, 3750–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.C. Multiresonant Coherent Multidimensional Spectroscopy. Annu. Rev. Phys. Chem. 2011, 62, 209–230. [Google Scholar]
- Wright, J.C.; Chen, P.C.; Hamilton, J.P.; Zilian, A.; LaBuda, M.J. Theoretical foundations for a new family of infrared four-wave mixing spectroscopies. Appl. Spectrosc. 1997, 51, 949–958. [Google Scholar] [CrossRef]
- Zhao, W.; Wright, J.C. Doubly vibrationally enhanced four wave mixing: The optical analog to 2D NMR. Phys. Rev. Lett. 2000, 84, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wright, J.C. Spectral simplification in vibrational spectroscopy using doubly vibrationally enhanced infrared four wave mixing. J. Am. Chem. Soc. 1999, 121, 10994–10998. [Google Scholar] [CrossRef]
- Donaldson, P.M.; Guo, R.; Fournier, F.; Gardner, E.M.; Barter, L.M.; Barnett, C.J.; Gould, I.R.; Klug, D.R.; Palmer, D.J.; Willison, K.R. Direct identification and decongestion of Fermi resonances by control of pulse time ordering in two-dimensional IR spectroscopy. J. Chem. Phys. 2007, 127, 114513. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.W. The Chemistry of Imines. Chem. Rev. 1963, 63, 489–510. [Google Scholar] [CrossRef]
- Jabin, I.; Revial, G.; Pfau, M.; Decroix, B.; Netchitaïlo, P. Tautomerism of α,β-Ethylenic Imines and Their Reactivity toward Electrophilic Olefins. Org. Lett. 1999, 1, 1901–1904. [Google Scholar] [CrossRef]
- Wei, Y.; Deb, I.; Yoshikai, N. Palladium-Catalyzed Aerobic Oxidative Cyclization of N-Aryl Imines: Indole Synthesis from Anilines and Ketones. J. Am. Chem. Soc. 2012, 134, 9098–9101. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Suri, M.; Glorius, F. Aerobic Synthesis of Pyrroles and Dihydropyrroles from Imines: Palladium(II)-Catalyzed Intramolecular C H Dehydrogenative Cyclization. Angew. Chem. Int. Ed. 2013, 52, 4892–4896. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.; Shang, Y.; Chen, Z.N.; Zhang, X.; Zhuang, W.; Su, W. Differentiation between enamines and tautomerizable imines in the oxidation reaction with TEMPO. Nat. Commun. 2018, 9, 5002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wright, J.C. Measurement of chi((3)) for doubly vibrationally enhanced four wave mixing spectroscopy. Phys. Rev. Lett. 1999, 83, 1950–1953. [Google Scholar] [CrossRef]
- Fournier, F.; Gardner, E.M.; Kedra, D.A.; Donaldson, P.M.; Guo, R.; Butcher, S.A.; Gould, I.R.; Willison, K.R.; Klug, D.R. Protein identification and quantification by two-dimensional infrared spectroscopy: Implications for an all-optical proteomic platform. Proc. Natl. Acad. Sci. USA 2008, 105, 15352–15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Fournier, F.; Donaldson, P.M.; Gardner, E.M.; Gould, I.R.; Klug, D.R. Detection of complex formation and determination of intermolecular geometry through electrical anharmonic coupling of molecular vibrations using electron-vibration-vibration two-dimensional infrared spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 8417–8421. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Mukamel, S.; Klug, D.R. Geometry determination of complexes in a molecular liquid mixture using electron-vibration-vibration two-dimensional infrared spectroscopy with a vibrational transition density cube method. Phys. Chem. Chem. Phys. 2012, 14, 14023–14033. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Long, D.A. Raman Spectroscopy; McGraw-Hill: New York, NY, USA, 1977. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.; Cha, S.; Cho, M.; Wright, J.C. Vibrational interactions of acetonitrile: Doubly vibrationally resonant IR–IR–visible four-wave-mixing spectroscopy. J. Chem. Phys. 2002, 117, 5675–5687. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Label | Coupling Modes/cm−1 | Vibrational Assignment1 | χmech | χelect | χ(3) |
|---|---|---|---|---|---|
| A | 1050.2/2030.7 | asymm CO stretch/ asymm CO stretch + CH2 wag (81%) | 1.51 × 10−15 | −2.28 × 10−16 | 1.28 × 10−15 |
| 1050.2/2047.8 | asymm CO stretch/ asymm CO stretch + CH3 wag (17%) | 6.70 × 10−16 | −2.07 × 10−16 | 4.63 × 10−16 | |
| B | 1050.2/2700.3 | asymm CO stretch/ asymm CO stretch + C=C stretch (97%) | 7.31 × 10−17 | −2.83 × 10−15 | −2.76 × 10−15 |
| C | 1334.0/2984.1 | CH rock/CH rock + C=C stretch (32%) | −1.56 × 10−16 | −7.42 × 10−16 | −8.97 × 10−16 |
| 1332.6/2982.7 | CH rock/CH rock + C=C stretch (28%) | −3.86 × 10−17 | −7.81 × 10−16 | −8.20 × 10−16 | |
| 1326.6/2976.7 | CH2 wag/CH2 wag + C=C stretch (26%) | −1.12 × 10−16 | −6.58 × 10−16 | −7.70 × 10−16 | |
| D | 1650.1/3300.2 | C=C stretch/C=C stretch + C=C stretch (100%) | 5.31 × 10−15 | −1.60 × 10−15 | 3.71 × 10−15 |
| Peak Label | Coupling Modes/cm−1 | Vibrational Assignment 1 | χmech | χelect | χ(3) |
|---|---|---|---|---|---|
| E | 1192.6/2385.2 | NH rock/NH rock + NH rock (100%) | −9.59 × 10−16 | −2.08 × 10−15 | −3.04 × 10−15 |
| F | 1192.6/2870.4 | NH rock/NH rock + C=C stretch | −5.14 × 10−18 | −4.96 × 10−15 | −4.97 ×10−15 |
| G | 1365.5/3043.3 | CH2 wag/CH2 wag + C=C stretch (100%) | −7.04 × 10−16 | −1.55 × 10−15 | −2.26 × 10−15 |
| H | 1677.8/2870.4 | C=C stretch/C=C stretch + NH rock (100%) | 1.45 × 10−17 | −1.86 × 10−15 | −1.85 × 10−15 |
| I | 1677.8/4556.6 | C=C stretch/C=C stretch + CH2 symm stretch (96%) | 1.65 × 10−16 | −2.02 × 10−15 | −1.86 × 10−15 |
| Peak Label | Coupling Modes /cm−1 | Vibrational Assignment 1 | χmech | χelect | χ(3) |
|---|---|---|---|---|---|
| J | 1074.4/4037.2 | C=N stretch/ C=N stretch + CH2 asymm stretch (31%) | 4.08 × 10−17 | −3.19 × 10−16 | −2.79 × 10−16 |
| 1074.4/4026.2 | C=N stretch/ C=N stretch + CH2 asymm stretch (24%) | −2.06 × 10−17 | −2.12 × 10−16 | −2.33 × 10−16 | |
| 1074.4/4022.4 | C=N stretch/ C=N stretch + CH2 asymm stretch (21%) | 3.35 × 10−17 | −2.51 × 10−16 | −2.17 × 10−16 | |
| 1091.8/4043.6 | CH2 twist/ CH2 twist + CH2 asymm stretch (16%) | −1.99 × 10−18 | −1.77 × 10−16 | −1.79 × 10−16 | |
| 1091.8/4054.5 | CH2 twist/ CH2 twist + CH2 asymm stretch (9%) | −6.30 × 10−17 | −5.93 × 10−17 | −1.22 × 10−16 | |
| K | 1723.5/2954.3 | C=N stretch/ C=N stretch + CH2 twist (100%) | 2.33 × 10−16 | 9.03 × 10−16 | 1.14 × 10−15 |
| L | 1723.5/3446.9 | C=N stretch/ C=N stretch + C=N stretch (100%) | 4.71 × 10−15 | 4.63 × 10−15 | 9.34 × 10−15 |
| Peak Label | Coupling Modes/cm−1 | Vibrational Assignment 1 | χmech | χelect | χ(3) |
|---|---|---|---|---|---|
| M | 1124.5/2820.0 | CH2 wag/ CH2 wag + C=C stretch (94%) | −3.87 × 10−16 | −3.89 × 10−15 | −4.28 × 10−15 |
| N | 1124.5/4427.1 | CH2 wag/ CH2 wag + NH stretch (96%) | −1.11× 10−16 | 2.36 × 10−15 | 2.25 × 10−15 |
| O | 1203.0/2898.5 | CH2 twist/ CH2 twist + C=C stretch (99%) | −2.79 × 10−16 | −1.90 × 10−15 | −2.18 × 10−15 |
| P | 1342.5/3038.1 | CH2 wag/ CH2 wag + C=C stretch (74%) | −1.29 × 10−16 | −1.25 × 10−15 | −1.38 × 10−15 |
| 1349.7/3045.3 | CH2 wag/ CH2 wag + C=C stretch (25%) | −3.78 × 10−17 | −6.12 × 10−16 | −6.50 × 10−16 | |
| Q | 1414.3/4717.0 | CH2 scissor/CH2 scissor + NH stretch (47%) | 2.97 × 10−16 | −3.47 × 10−15 | −3.18 × 10−15 |
| 1426.4/4729.0 | CH2 scissor/CH2 scissor +NH stretch (23%) | 2.27 × 10−16 | −2.10 × 10−15 | −1.88 × 10−15 | |
| 1419.3/4721.9 | CH2 scissor/CH2 scissor +NH stretch (22%) | 2.64 × 10−16 | −2.10 × 10−15 | −1.83 × 10−15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, F.; Chen, Z.; Han, K.; Zhang, L.; Zhuang, W. Differentiation between Enamines and Tautomerizable Imines Oxidation Reaction Mechanism using Electron-Vibration-Vibration Two Dimensional Infrared Spectroscopy. Molecules 2019, 24, 869. https://doi.org/10.3390/molecules24050869
Long F, Chen Z, Han K, Zhang L, Zhuang W. Differentiation between Enamines and Tautomerizable Imines Oxidation Reaction Mechanism using Electron-Vibration-Vibration Two Dimensional Infrared Spectroscopy. Molecules. 2019; 24(5):869. https://doi.org/10.3390/molecules24050869
Chicago/Turabian StyleLong, Fengqin, Zheng Chen, Keli Han, Lu Zhang, and Wei Zhuang. 2019. "Differentiation between Enamines and Tautomerizable Imines Oxidation Reaction Mechanism using Electron-Vibration-Vibration Two Dimensional Infrared Spectroscopy" Molecules 24, no. 5: 869. https://doi.org/10.3390/molecules24050869
APA StyleLong, F., Chen, Z., Han, K., Zhang, L., & Zhuang, W. (2019). Differentiation between Enamines and Tautomerizable Imines Oxidation Reaction Mechanism using Electron-Vibration-Vibration Two Dimensional Infrared Spectroscopy. Molecules, 24(5), 869. https://doi.org/10.3390/molecules24050869