Quantitative Characterization of Olaparib in Nanodelivery System and Target Cell Compartments by LC-MS/MS

 , ,
, , 

Abstract
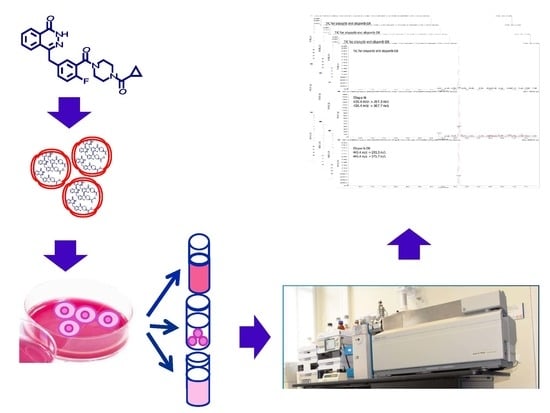
:
1. Introduction
2. Results
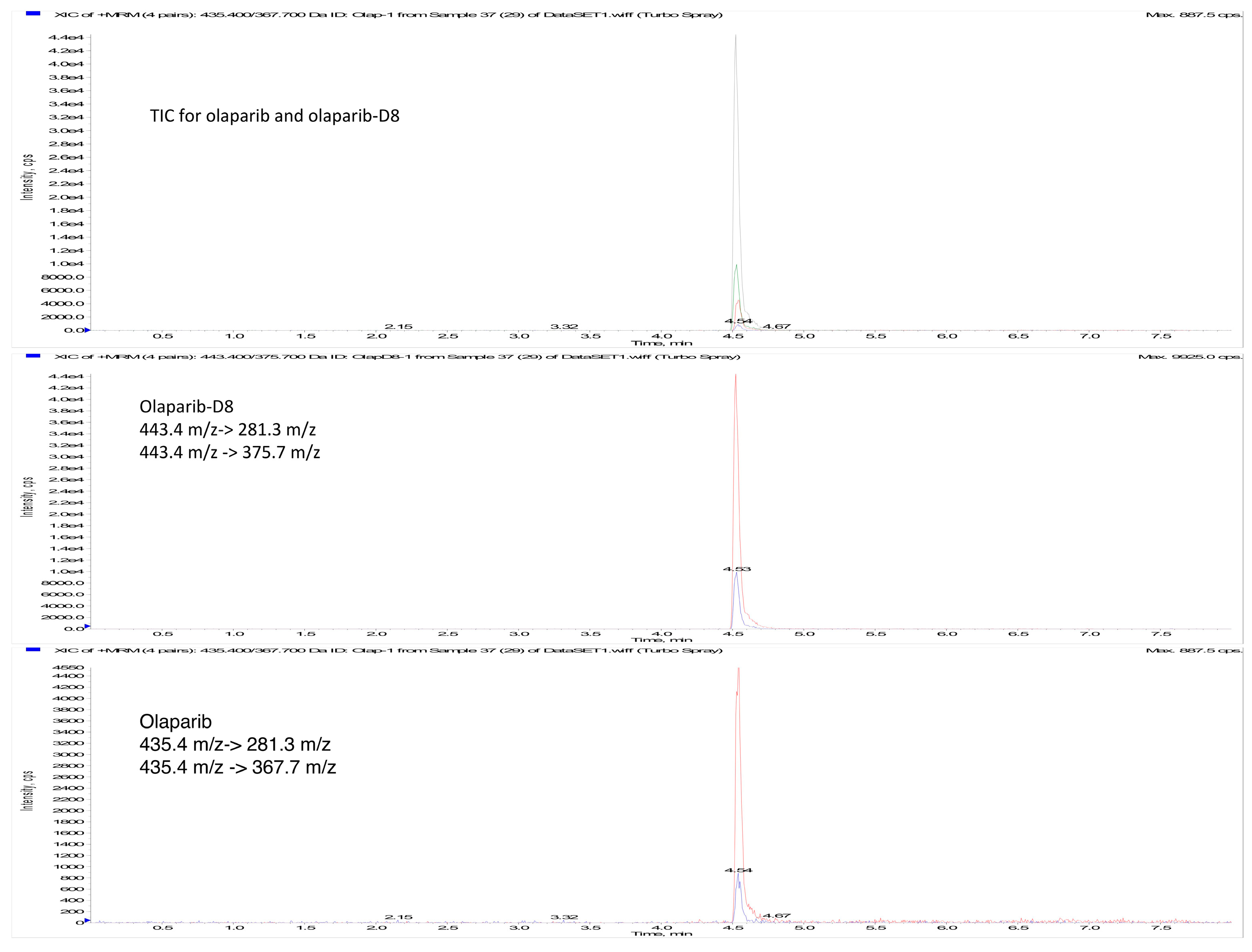
2.1. Instrument Parameters
2.2. Sample Preparation
2.3. Method Validation
2.3.1. Linearity, LOD and LOQ
2.3.2. Precision and Accuracy
2.3.3. Recovery and Matrix Effect
2.3.4. Stability
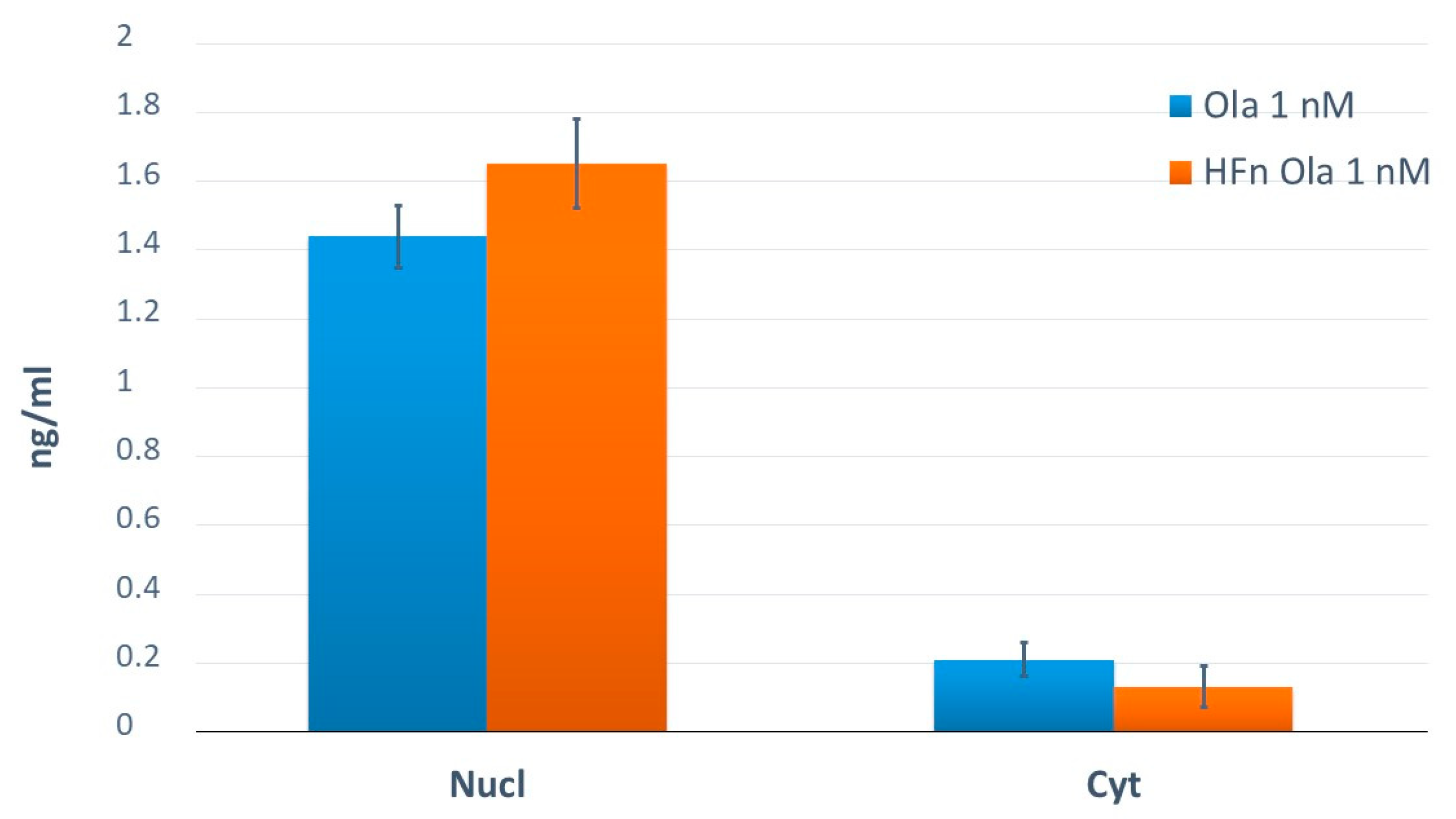
2.4. Application of the Method
3. Discussion
3.1. Sample Preparation
3.2. Method Validation
4. Materials and Methods
4.1. Chemicals
4.2. Instruments
4.3. Standard Solutions
4.4. Control Plasma, Urine, and Mouse Tissue, Cell Extracts and Ola Loading Samples Collection
4.4.1. Ola Loading Samples
4.4.2. Cells Samples
4.4.3. Plasma and Urines
4.4.4. Tissue Samples
4.5. Calibration Curves
4.5.1. Ola Loading Samples
4.5.2. Cell Nuclei Samples
4.5.3. Culture Medium and Cytoplasmic Fraction
4.5.4. Plasma and Urine Samples
4.5.5. Tissue Samples
4.5.6. Quality Control (QC) Samples
4.6. Sample Preparation
4.6.1. Ola Loading Samples
4.6.2. Cell Nuclei Samples
4.6.3. Culture Medium, Cytoplasmic Fraction and Tissue Samples
4.6.4. Plasma and Urine Samples
4.7. Sample Preparation
4.7.1. Sensitivity and Carry-Over
4.7.2. Linearity, Precision, and Accuracy
4.7.3. Recovery and Matrix Effect
4.7.4. Sample Stability
4.7.5. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellini, M.; Mazzucchelli, S.; Galbiati, E.; Sommaruga, S.; Fiandra, L.; Truffi, M.; Rizzuto, M.A.; Colombo, M.; Tortora, P.; Corsi, F.; et al. Protein nanocages for self-triggered nuclear delivery of DNA-targeted chemotherapeutics in Cancer Cells. J. Control Release 2014, 196, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Swaisland, H.; Lau, A.; O’Connor, M.J.; Ashworth, A.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nokihara, H.; Yamada, Y.; Goto, Y.; Tanioka, M.; Shibata, T.; Yamada, K.; Asahina, H.; Kawata, T.; Shi, X.; et al. A Phase I, dose-finding and pharmacokinetic study of olaparib (AZD2281) in Japanese patients with advanced solid tumors. Cancer Sci. 2012, 103, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Bundred, N.; Gardovskis, J.; Jaskiewicz, J.; Eglitis, J.; Paramonov, V.; McCormack, P.; Swaisland, H.; Cavallin, M.; Parry, T.; Carmichael, J.; et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: A Phase I multicentre trial in patients scheduled for elective breast cancer surgery. Investig. New Drugs 2013, 31, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, C.M.; Lucas, L.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Development and validation of a high-performance liquid chromatography-tandem mass spectrometry assay quantifying olaparib in human plasma. J. Chromatogr. B 2013, 940, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sparidans, R.W.; Martens, I.; Valkenburg-van Iersel, L.B.J.; den Hartigh, J.; Schelleris, J.H.M.; Beijnen, J.H. Liquid chromatography-tandem mass spectrometric assay for the PARP-1 inhibitor olaparib in combination with the nitrogen mustard melphalan in human plasma. J. Chromatogr. B 2011, 879, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Swaisland, H.; Leunen, K.; Rutten, A.; Soetekouw, P.; Slater, S.; Verheul, H.M.; Fielding, A.; So, K.; Bannister, W.; et al. Effect of Food on the Pharmacokinetics of Olaparib after Oral Dosing of the Capsule Formulation in Patients with Advanced Solid Tumors. Adv. Ther. 2015, 32, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Peer, C.J.; Mannargudi, B.; Swaisland, H.; Lee, J.-M.; Kohn, E.C.; Figg, W.D. A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application. Chromatography 2014, 1, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Daumar, P.; Dufour, R.; Dubois, C.; Penault-Llorca, F.; Bamdad, M.; Mounetou, E. Development and validation of a high-performance liquid chromatography method for the quantitation of intracellular PARP inhibitor Olaparib in cancer cells. J. Pharm. Biomed. Anal. 2018, 152, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, S.; Truffi, M.; Baccarini, F.; Beretta, M.; Sorrentino, L.; Bellini, M.; Rizzuto, M.A.; Ottria, R.; Ravelli, A.; Ciuffreda, P.; et al. H-Ferritin-nanocaged olaparib: A promising choice for both BRCA mutated and sporadic triple negative breast cancer. Sci. Rep. 2017, 7, 7505–7520. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry, Bioanalytical Method Validation. Guidance for Industry: Bioanalytical Method Validations. 2001. Available online: http://www.fda.ov/downloads/Drugs/Guidances/ucm070107 (accessed on 21 September 2018).
- Mazzucchelli, S.; Ravelli, A.; Gigli, F.; Minoli, M.; Corsi, F.; Ciuffreda, P.; Ottria, R. LC-MS/MS method development for quantification of doxorubicin and its metabolite 13-hydroxy doxorubicin in mice biological matrices: Application to a pharmaco-delivery study. Biomed. Chrom. 2017, 31, e3863. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds Ola and Olad8 are available from the authors. Samples of loading compounds are not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix Effect | Recovery | ||||||
|---|---|---|---|---|---|---|---|
| Matrix | LOD | LOQ | LQC | HQC | LQC | MQC | HQC |
| Cytoplasmic Fraction | 0.04 | 0.10 | 36 | 31 | 88 | 87 | 90 |
| Nuclei Pellet | 0.21 | 0.48 | −9 | −17 | 85 | 88 | 92 |
| Phosphate Buffer | 0.02 | 0.09 | 5 | 11 | 91 | 96 | 93 |
| Plasma | 0.19 | 0.48 | −15 | −21 | 75 | 81 | 80 |
| Urine | 0.02 | 0.12 | 7 | 3 | 89 | 92 | 97 |
| Liver | 0.47 | 1.54 | 26 | 31 | 58 | 75 | 80 |
| Kidney | 0.86 | 2.87 | 32 | 28 | 63 | 82 | 85 |
| (%CV) Intra-Day | (%CV) Inter-Day | (%RSE) Intra-Day | (%RSE) Inter-Day | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MATRICES | LQC | MQC | HQC | LQC | MQC | HQC | LQC | MQC | HQC | LQC | MQC | HQC |
| Phosphate Buffer | 5.2 | 4.7 | 6.8 | 7.6 | 9.8 | 4.7 | 4.8 | 5.4 | 6.9 | 9.1 | 12.2 | 9.9 |
| Cytoplasmic Fraction | 5.6 | 7.6 | 4.3 | 8.8 | 6.6 | 5.4 | 8.6 | 7.1 | 7.2 | 11.7 | 11.3 | 9.8 |
| Nuclei Pellet | 4.4 | 3.2 | 6.1 | 4.7 | 6.6 | 5.8 | 11.1 | 6.3 | 8.1 | 12.2 | 8.5 | 7.5 |
| Plasma | 7.7 | 8.2 | 5.4 | 9.5 | 7.4 | 8.6 | 10.4 | 8.9 | 8.8 | 13.4 | 9.7 | 10.2 |
| Urine | 3.7 | 2.7 | 5.2 | 4.7 | 7.5 | 7.3 | 6.9 | 4.8 | 5.5 | 9.8 | 8.1 | 6.9 |
| Liver | 5.4 | 7.6 | 3.7 | 7.3 | 6.7 | 5.9 | 9.4 | 7.5 | 6.6 | 9.6 | 8.5 | 7.8 |
| Kidney | 8.7 | 6.2 | 5.7 | 6.2 | 6.5 | 7.3 | 8.9 | 8.1 | 8.9 | 11.2 | 11.8 | 10.8 |
| LQC2 | MQC2 | HQC2 | LQC2 | MQC2 | HQC2 | LQC2 | MQC2 | HQC2 | LQC2 | MQC2 | HQC2 | |
| Plasma | 9.9 | 8.1 | 6.7 | 10.1 | 8.1 | 8.9 | 10.1 | 9.9 | 8.5 | 13.8 | 10.4 | 9.9 |
| Urine | 4.8 | 5.2 | 6.1 | 5.5 | 6.8 | 6.5 | 6.7 | 6.8 | 7.8 | 10.5 | 8.5 | 6.7 |
| Loading | 5.7 | 5.2 | 2.3 | 4.5 | 4.3 | 4.5 | 3.6 | 4.6 | 2.9 | 5.1 | 3.8 | 4.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottria, R.; Ravelli, A.; Miceli, M.; Casati, S.; Orioli, M.; Ciuffreda, P. Quantitative Characterization of Olaparib in Nanodelivery System and Target Cell Compartments by LC-MS/MS. Molecules 2019, 24, 989. https://doi.org/10.3390/molecules24050989
Ottria R, Ravelli A, Miceli M, Casati S, Orioli M, Ciuffreda P. Quantitative Characterization of Olaparib in Nanodelivery System and Target Cell Compartments by LC-MS/MS. Molecules. 2019; 24(5):989. https://doi.org/10.3390/molecules24050989
Chicago/Turabian StyleOttria, Roberta, Alessandro Ravelli, Matteo Miceli, Sara Casati, Marica Orioli, and Pierangela Ciuffreda. 2019. "Quantitative Characterization of Olaparib in Nanodelivery System and Target Cell Compartments by LC-MS/MS" Molecules 24, no. 5: 989. https://doi.org/10.3390/molecules24050989
APA StyleOttria, R., Ravelli, A., Miceli, M., Casati, S., Orioli, M., & Ciuffreda, P. (2019). Quantitative Characterization of Olaparib in Nanodelivery System and Target Cell Compartments by LC-MS/MS. Molecules, 24(5), 989. https://doi.org/10.3390/molecules24050989