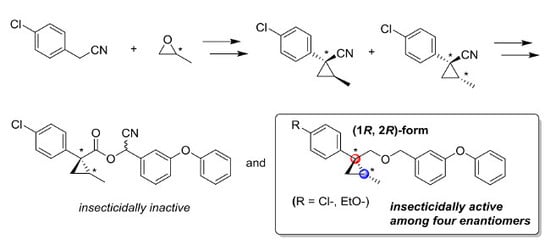
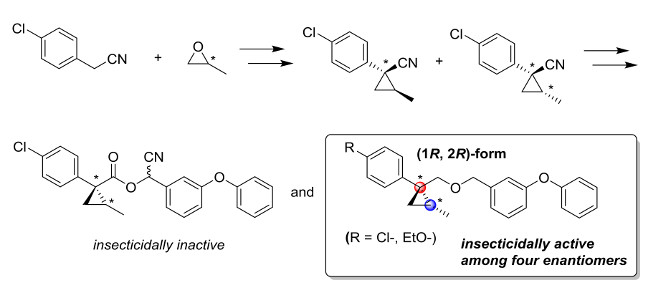
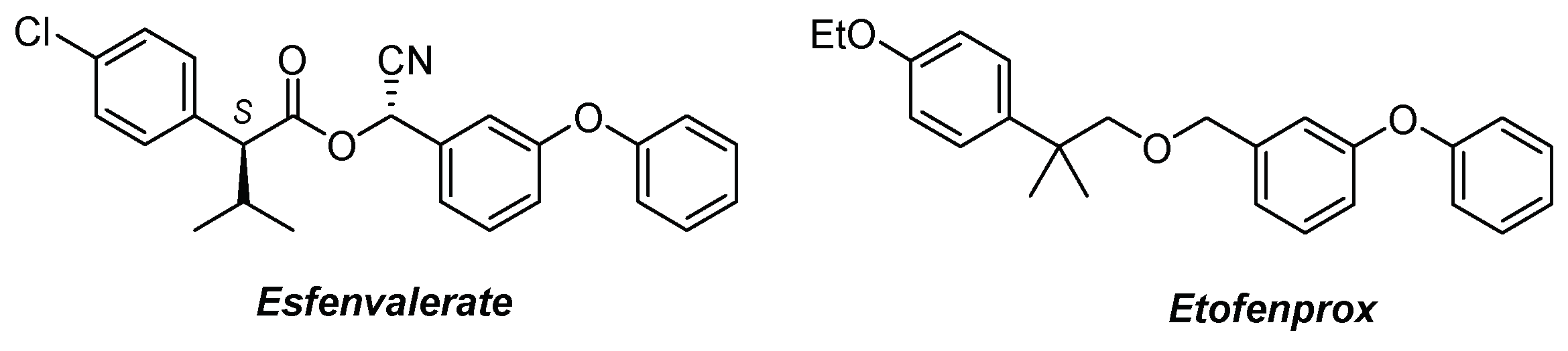
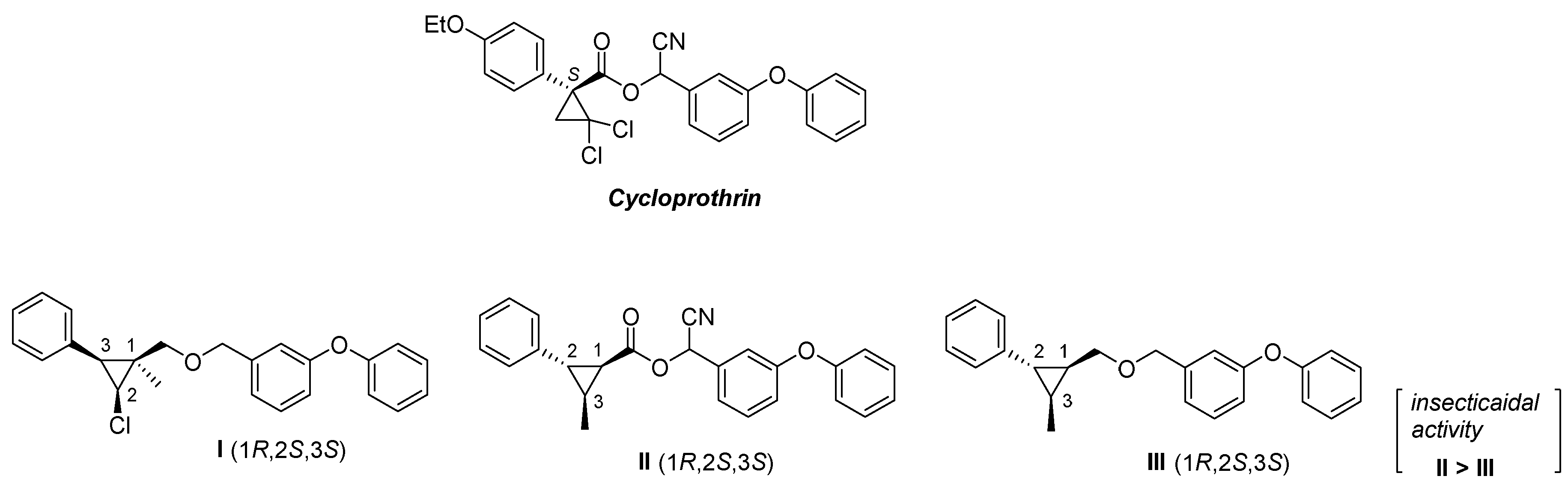
3. Materials and Methods
All reactions were carried out in oven-dried glassware under an argon atmosphere. Flash column chromatography was performed with silica gel 60 (230–400 mesh ASTM, Merck, Darmstat, Germany). TLC analysis was performed on Merck 0.25 mm Silicagel 60 F
254 plates. Melting points were determined on a hot stage microscope apparatus (ATM-01, AS ONE, Osaka, Japan) and were uncorrected. NMR spectra were recorded on a JEOLRESONANCE EXC-400 or ECX-500 spectrometer (JEOL, Akishima, Japan) operating at 400 MHz or 500 MHz for
1H-NMR, and 100 MHz and 125 MHz for
13C-NMR. Chemical shifts (δ ppm) in CDCl
3 were reported downfield from TMS (= 0) for
1H-NMR. For
13C-NMR, chemical shifts were reported in the scale relative to CDCl
3 (77.00 ppm) as an internal reference. IR Spectra were recorded on FT/IR-5300 spectrophotometer (JASCO, Akishima, Japan). Mass spectra were measured on a JMS-T100LC spectrometer (JEOL). HPLC data were obtained on a SHIMADZU (Kyoto, Japan) HPLC system (consisting of the following: LC-20AT, CMB20A, CTO-20AC, and detector SPD-20A measured at 254 nm) using Chiracel AD-H or Ad-3 column (Daicel, Himeji, Japan, 25 cm) at 25 °C. Optical rotations were measured on a JASCO DIP-370 (Na lamp, 589 nm). The synthetic procedure of
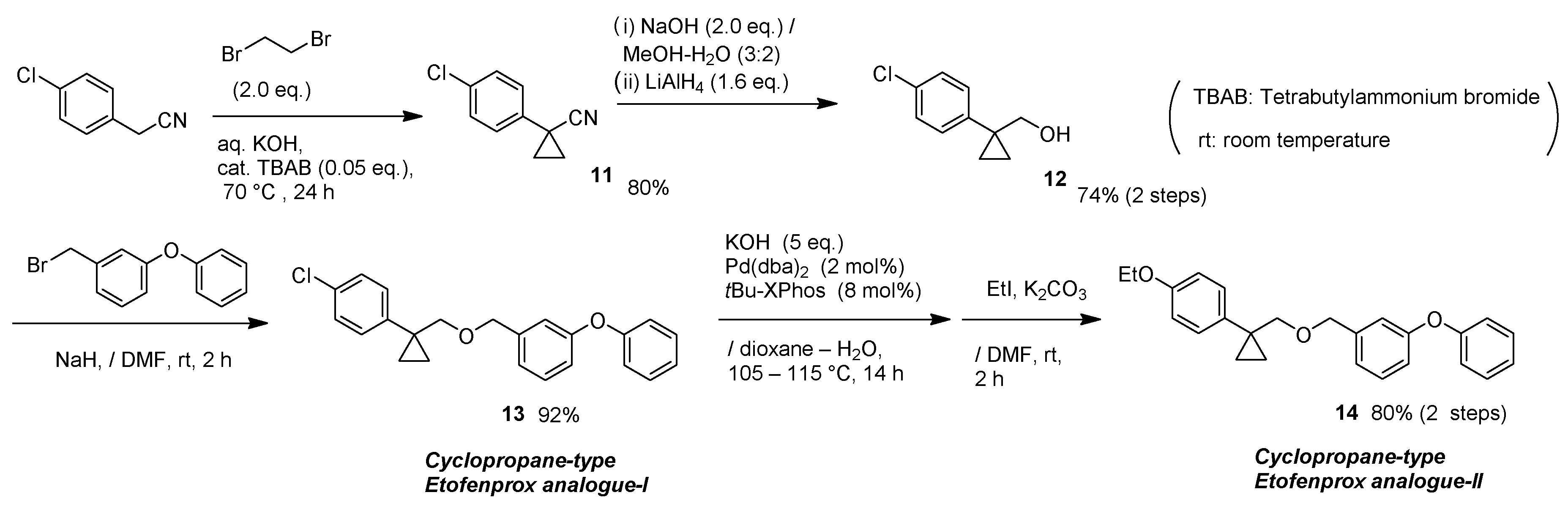
11,
12,
13,
14,
16,
17,
18, and etofenprox are available in the
supplementary information.
1H-,
13C-NMR spectra for compounds (±)-
5, (±)-
3, (
1R*,
2S*)-
4, (
1R*,2R*)-
4, (
1R*,
2S*)-
6, (
1R*,
2R*)-
6, (
1R*,
2S*)-
8, (
1R*,
2R*)-
8, (
1R*,
2S*)-
7, (
1R*,
2R*)-
7, (
1R*,
2S*)-
9, (
1R*,
2R*)-
9, (
1R,
2R)-
10 can see in the
supplementary information.
(±)-2-(4-Chlorophenyl)-4-hydroxypentanenitrile: (±)-5 (diastereo mixtures)
![Molecules 24 01023 i001]()
LHMDS (1.1 M in THF, 10 mL, 11 mmol) was added to a stirred suspension of 4-chlorobenzyl cyanide (1.52 g, 10 mmol) in THF (13 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 1 h. (±)-Propylene oxide (2.1 mL, 30 mmol) was added to the mixture, which was stirred for 1 h. The mixture was quenched with 1M-HCl aq., which was extracted three times with AcOEt. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/AcOEt = 10:1 to 1:2) to give the desired product (±)-3 (1.48 g, 70%, dr = 65:35). Pale yellow oil; 1H-NMR (400 MHz, CDCl3) δ = 1.23 (d, J = 6.4 Hz, 3H × 7/20), 1.29 (d, J = 6.0 Hz, 3H × 13/20), 1.50–1.70 (brs, 1H), 1.79–1.86 (m, 1H × 13/20), 1.89–2.01 (m, 1H), 2.10–2.17 (m, 1H × 7/20), 3.56–3.67 (m, 1H × 13/20), 4.04 (dd, J = 5.5 Hz, J = 9.6 Hz, 1H × 7/20), 4.14–4.21 (m, 1H), 7.28–7.38 (m, 4H); 13C-NMR (125 MHz; CDCl3) δ = 23.6, 23.8, 32.7, 33.7, 43.8, 44.7, 63.9, 65.0, 120.4, 121.1, 128.4, 129.0, 129.2 (2C), 133.7, 133.8, 134.0, 134.4; νmax (neat)/cm−1 3424, 2968, 2241, 1493, 1410, 1375, 1130, 1093, 1014, 953, 932, 845, 822, 791, 779, 718; HRMS (DART) calcd for C11H12ClNO [M + H]+ 210.0686, found 210.0688.
(4R)-2-(4-Chlorophenyl)-4-hydroxypentanenitrile: (R)-5 (diastereo mixtures)
Following the similar procedure for the preparation of (±)-3, the reaction of chlorobenzyl cyanide (758 mg, 5.0 mmol), (R)-propylene oxide (1.05 mL, 15 mmol), and LHMDS (1.1 M in THF, 5.0 mL, 5.5 mL), and the successive SiO2-gel column chromatographic purification gave the desired product (R)-5 (813 mg, 78%, dr = 65:35). Pale yellow oil.
(4S)-2-(4-Chlorophenyl)-4-hydroxypentanenitrile: (S)-5 (diastereo mixtures)
Following the similar procedure for the preparation of (±)-3, the reaction of chlorobenzyl cyanide (1.52 g, 10 mmol), (S)-propylene oxide (2.1 mL, 30 mmol) and LHMDS (1.1 M in THF, 8.46 mL, 1.1 mL), and the successive SiO2-gel column chromatographic purification gave the desired product (S)-5 (1.57 g, 75%, dr = 65:35). Pale yellow oil.
(±)-4-(4-Chlorophenyl)-4-cyanobutan-2-yl 4-methylbenzenesulfonate: (±)-3 (diastereo mixtures)
![Molecules 24 01023 i004]()
TsC1 (286 mg, 1.5 mmol) in MeCN (1.0 mL) was added to a stirred solution of (±)-3 (210 mg, 1.0 mmol, dr = 65:35), Et3N (202 mg, 2.0 mmol), and Me3N·HC1 (10 mg, 0.1 mmol) in MeCN (1.0 mL) at 0–5 °C, and the mixture was stirred for 1 h. Water was added to the mixture, which was extracted twice with EtOAc. The combined organic phase was washed with water and brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/AcOEt = 30:1 to 10:1) to give the desired product (±)-3 (350 mg, 96%, dr = 65:35). Pale yellow oil; 1H-NMR (400 MHz, CDCl3) δ = 1.27 (d, J = 6.4 Hz, 3H × 7/20), 1.32 (d, J = 6.4 Hz, 3H × 13/20), 1.99–2.11 (m, 2H,), 2.47 (s, 3H), 3.81 (dd, J = 5.5 Hz, J = 10.1 Hz, 1H × 13/20), 3.90 (dd, J = 5.5 Hz, J = 8.7 Hz, 1H × 7/20), 4.51–4.59 (m, 1H × 7/20), 4.77–4.85 (m, 1H × 13/20), 7.20–7.40 (m, 6H), 7.77–7.79 (m, 2H × 7/20), 7.85–7.87 (m, 2H × 13/20); 13C-NMR (125 MHz; CDCl3) δ = 20.3, 20.6, 21.3 (2C), 31.9, 33.0, 41.4, 42.3, 75.7, 76.6, 119.1, 119.8, 127.3, 127.5, 128.2, 128.8, 129.0, 129.1, 129.7, 129.8, 132.8, 133.1, 133.2, 133.4, 133.8, 134.0, 144.8, 145.0.; νmax (neat)/cm−1 3022, 2984, 2243, 1597, 1493, 1362, 1188, 1175, 1096, 1059, 1016, 924, 891, 816, 750, 687, 662.
(2R)-4-(4-Chlorophenyl)-4-cyanobutan-2-yl 4-methylbenzenesulfonate: (R)-3 (diastereo mixtures)
Following the similar procedure for the preparation of (±)-3, the reaction of (R)-5 (629 mg, 3.0 mmol, dr = 50:50), TsCl (858 mg, 4.5 mmol), Et3N (607 mg, 6.0 mmol), and Me3N·HCl (29 mg, 0.3 mmol), and the successive SiO2-gel column chromatographic purification gave the desired product (R)-3 (980 mg, 90%, dr = 65:35). Pale yellow oil.
(2S)-4-(4-Chlorophenyl)-4-cyanobutan-2-yl 4-methylbenzenesulfonate: (S)-3 (diastereo mixtures)
Following the similar procedure for the preparation of (±)-3, the reaction of (S)-5 (1.54 g, 7.34 mmol, dr = 50:50), TsCl (2.10 g, 11.0 mmol), Et3N (1.49 g, 14.7 mmol), and Me3N·HCl (70 mg, 0.7 mmol), and the successive SiO2-gel column chromatographic purification gave the desired product (S)-3 (2.45 g, 92%, dr = 65:35). Pale yellow oil.
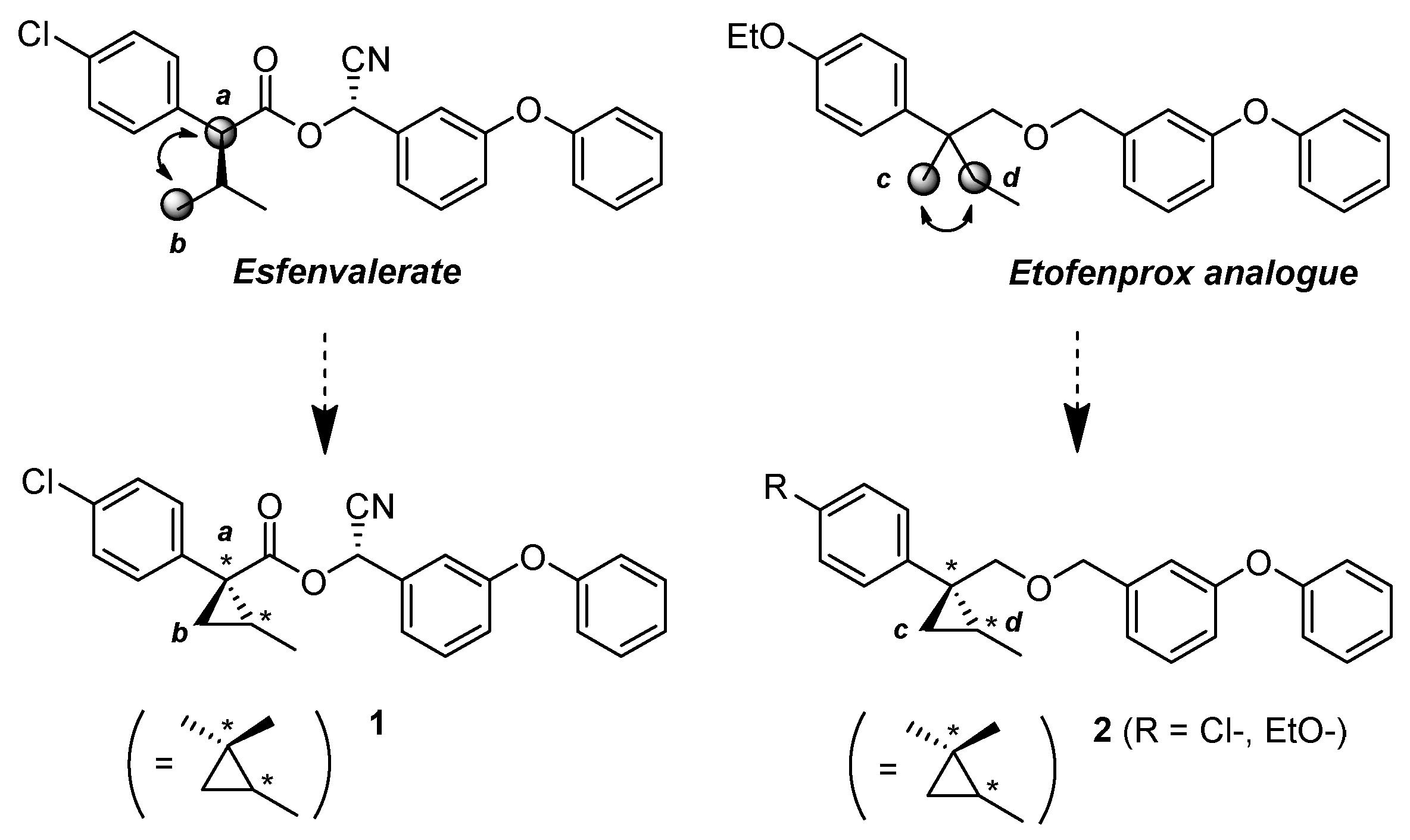
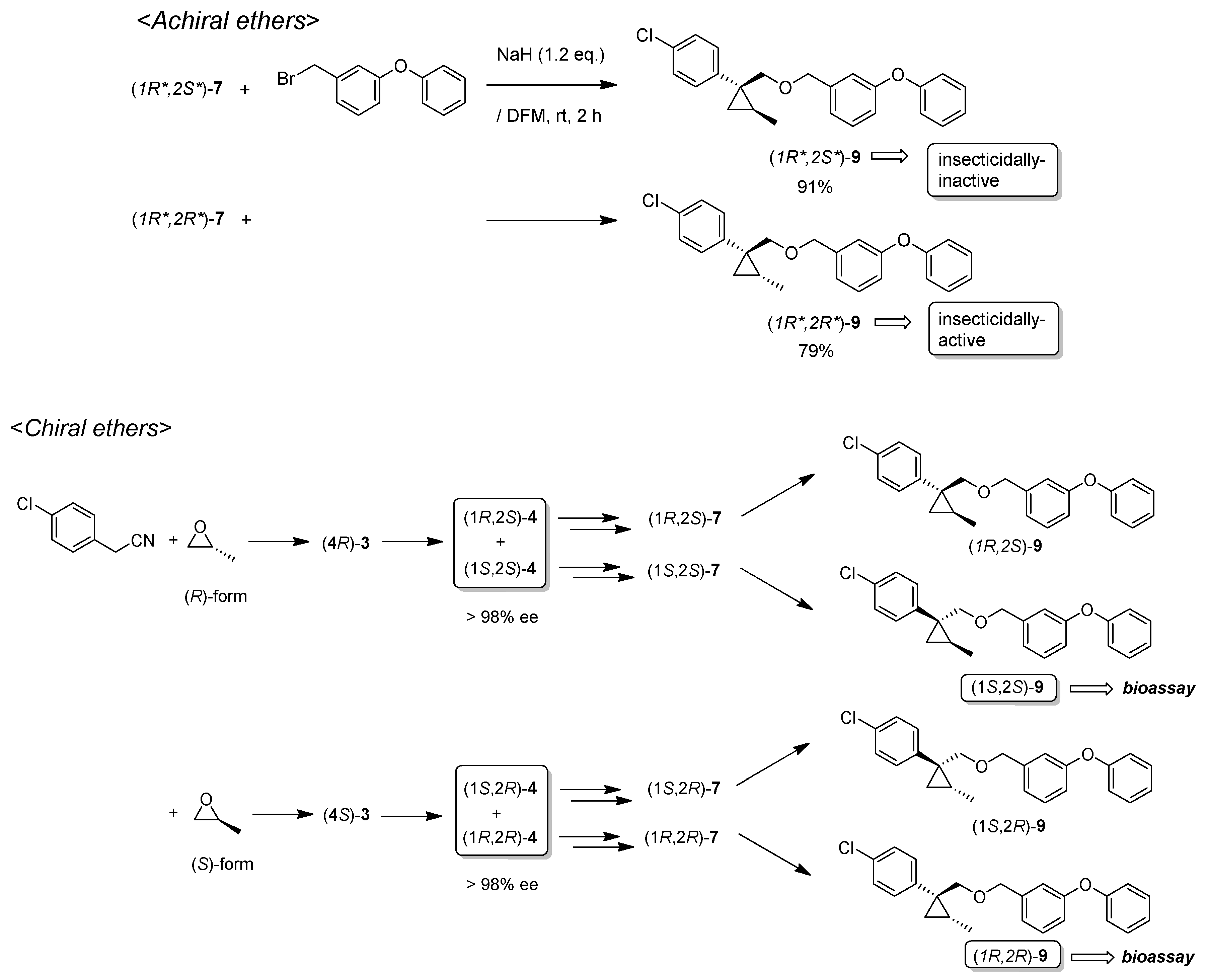
(1R*,2S*)- and (1R*,2R*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carbonitrile: (1R*,2S*)-4 and (1R*,2R*)-4
Tosylate (±)-3 (65:35: diastereo mixtures) (942 mg, 2.59 mmol) in Et2O (13 mL) was added to a stirred suspension of tBuOLi (249 mg, 3.11 mmol) in Et2O (13 mL) at −78 °C under Ar atmosphere, followed by being stirred at the same temperature for 0.5 h. The mixture was allowed to warm to 0–5 °C, and was stirred for 2 h. The mixture was quenched with water, which was extracted three times with AcOEt. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/AcOEt = 50:1) to give (1R*,2S*)-4 (175mg, 35%) and (1R*,2R*)-4 (161 mg, 32%).
(1R*,2S*)-4: Colorless oil; 1H-NMR (400 MHz, CDCl3) δ = 1.41 (dd, J = 10.1 Hz, J = 11.9 Hz, 1H), 1.47–1.49 (m, 3H), 1.56–1.60 (m, 2H), 7.18–7.21 (m, 2H), 7.29–7.33 (m, 2H); 13C-NMR (125 MHz, CDCl3): δ 15.9, 20.0, 25.3(2C), 120.4, 126.8, 128.8, 133.1, 135.2.; νmax (neat)/cm−1 2968, 2934, 2234, 1495, 1445, 1402, 1369, 1121, 1096, 1015, 889, 849, 827, 764, 727.; HRMS (DART) calcd for C11H10OClN [M + H]+ 192.0580, found 192.0573.
(1R*,2R*)-4: Colorless oil; 1H-NMR (400 MHz, CDCl3) δ = 0.83 (d, J = 6.5 Hz, 3H), 1.26 (dd, J = 5.5 Hz, J = 6.9 Hz, 1H), 1.74 (dd, J = 5.5 Hz, J = 9.3 Hz, 1H), 1.83–1.94 (m, 1H), 7.25–7.38 (m, 4H); 13C-NMR (125 MHz; CDCl3): δ 13.6, 17.4, 19.9, 23.2, 123.2, 128.9, 130.7, 131.0, 134.1.; νmax (neat)/cm−1 2966, 2230, 1493, 1445, 1396, 1366, 1315, 1124, 1094, 1043, 1014, 997, 908, 862, 827, 789, 729.
(1R,2S)- and (1S,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carbonitrile: (1R,2S)-4 and (1S,2S)-4
Following the similar cyclization procedure described abave, (1R,2S)-4 (45%) and (1S,2S)-4 (39%) were prepared from (2R)-3.
(1R,2S)-4: Colorless oil; [α] +64.9 (c 1.00, CHCl3); 98% ee, HPLC analysis (Daicel Chiralcel OD-H column, solvent: hexane/ethanol = 150:1, flow rate 0.80 mL/min, 254 nm UV detector), tR(racemate) = 10.38 min and 11.18 min. tR[(1R,2S)-form] = 9.74 min
(1S,2S)-4: White colored solid; m.p. 73–75 °C, [α] +36.6 (c 0.7, CHCl3); >98% ee, HPLC analysis (Daicel Chiralcel OD-H column, solvent: hexane/ethanol = 300:1, flow rate 0.80 mL/min, 254 nm UV detector), tR(racemate) = 17.41 min and 18.55 min. tR[(1S,2S)-form] = 19.01 min.
(1S,2R) and (1R,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carbonitrile: (1S,2R)-4, (1R,2R)-4
Following the similar cyclization procedure described above, (1S,2R)-4 (40%) and (1R,2R)-4 (38%) were prepared from (2S)-3.
(1S,2R)-3: Colorless oil; [α] −62.6 (c 0.25, CHCl3); 98% ee, HPLC analysis (Daicel Chiralcel OD-H column, solvent: hexane/ethanol = 150:1, flow rate 0.80 mL/min, 254 nm UV detector), tR(racemate) = 10.38 min and 11.18 min. tR[(1S,2R)-form] = 11.36 min.
(1R,2R)-4: White colored solid; m.p. 73-74 °C; [α] −33.4 (c 0.33, CHCl3); >98% ee, HPLC analysis (Daicel Chiralcel OD-H column, solvent: hexane/ethanol = 300:1, flow rate 0.80 mL/min, 254 nm UV detector), tR(racemate) = 17.41 min and 18.55 min. tR[(1R,2R)-form] = 15.01 min.
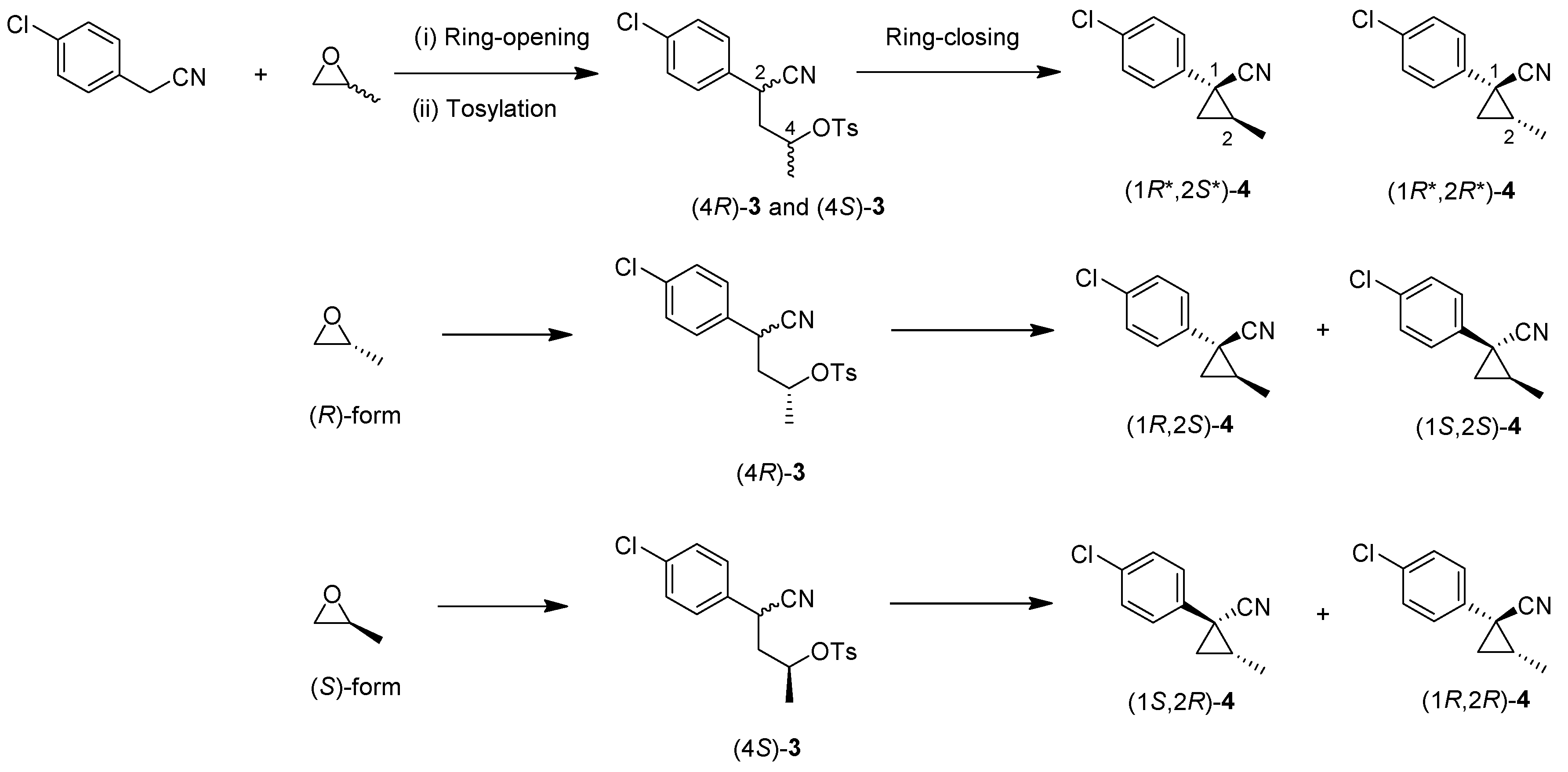
(1R*,2S*)-1-(4-chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1R*,2S*)-6
![Molecules 24 01023 i010]()
DIBAL (1.01 M in toluene, 1.98 mL, 2.0 mmol) was added slowly to a stirred solution of (1R*,2S*)-4 (192 mg, 1.0 mmol) in toluene (10 mL) at −78 °C under an Ar atmosphere. The mixture was allowed to warm to 20–25 °C over 1 h, and then the mixture was quenched with MeOH. Then, potassium sodium tartrate tetrahydrate solution was added to the mixture. After stirring for 0.5 h, the mixture was extracted three times with EtOAc. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. The obtained crude aldehyde product (140 mg) was used for the next step without purification. To a stirred solution of the obtained aldehyde (140 mg), 2-methyl-2-butene (561 mg, 8.0 mmol) in tBuOH (9 mL), water (4 mL) were added NaH2PO4 (480 mg, 4.0 mmol) in water (4 mL) and NaClO2 (217 mg, 2.4 mmol) in water (4 mL) at 20–25 °C, and the mixture was stirred at that temperature for 1 h. The reaction mixture was quenched with water and extracted with AcOEt three times. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated. Sat. NaHCO3 aq. was added to the residue, which was wasehd with Et2O two times. The separated aqueous phase was acidified with 1M HCl aq. solution, which was extracted with AcOEt three times. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated to give the desired product (1R*,2S*)-6 (160 mg, 76% for two steps). White colored solid; m.p. 136–137 °C; 1H-NMR (400 MHz, CDCl3) δ = 1.33 (dd, J = 4.5 Hz, 8.9 Hz, 1H), 1.37 (d, J = 6.2 Hz, 3H), 1.56 (dd, J = 4.5 Hz, 7.6 Hz, 1H), 1.59–1.69 (m, 1H), 7.26–7.27 (m, 4H); 13C-NMR (125 MHz; CDCl3): δ 12.7, 22.6, 25.3, 33.7, 128.3, 131.6, 133.0, 139.0, 178.6.; νmax (neat)/cm−1 3011, 2880, 2598, 2363, 1686, 1495, 1420, 1312, 1213, 1092, 1013, 889, 829, 754, 719.
(1R,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1R,2S)-6
Following the similar DIDAL-reduction and Pinnick oxidation procedure described above, (1R,2S)-6 was prepared from (1R,2S)-4 (80%, two steps). White colored crystal; m.p. 104–106 °C; [α] +120.6 (c 0.16, CHCl3).
(1S,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1S,2R)-6
Following the similar DIDAL-reduction and Pinnick oxidation procedure described above, (1S,2R)-6 (79%, 2 steps) was prepared from (1S,2R)-4 (79%, two steps). White colored crystals; m.p. 102–105 °C; [α] −116.4 (c 0.97, CHCl3).
(1R*,2R*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1R*,2R*)-6
![Molecules 24 01023 i013]()
A mixture of (1R*,2R*)-4 (381 mg, 2.0 mmol) and NaOH (1.6 g, 40 mmol) in MeOH (10.0 mL) and H2O (6.6 mL) was heated at 100 °C for 16 h. After cooling down, the mixture was quenched with water, which was washed with Et2O two times. The separated aqueous phase was acidified with 1M HCl aq. solution, which was extracted with EtOAc three times. The combined organic phase was washed with brine, dried (Na2SO4) and concentrated to give the desired product (1R*,2R*)-6 (358 mg, 84%). White colored crystals: m.p. 130–133 °C; 1H-NMR (400 MHz, CDCl3) δ = 0.83 (d, J = 6.4 Hz, 3H), 1.09 (dd, J = 4.6 Hz, J = 6.9 Hz, 1H), 1.80 (dd, J = 4.6 Hz, J = 9.2 Hz, 1H), 1.90–1.98 (m, 1H), 7.18–7.22 (m, 2H), 7.28–7.32 (m, 2H); 13C-NMR (125 MHz; CDCl3): δ 12.7, 22.6, 25.3, 33.7, 128.3, 131.6, 133.0, 139.0, 178.6.; νmax (neat)/cm−1 3011, 2880, 2598, 2363, 1686, 1495, 1420, 1312, 1213, 1092, 1013, 943, 889, 829, 754, 719; HRMS (DART) calcd for C11H11ClO2 [M + H]+ 211.0526, found 211.0523.
(1R,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1R,2R)-6
Following the similar hydroxylation procedure described above, (1S,2S)-6 was prepared from (1S,2S)-4 (92%). White colored crystals; m.p. 103–104 °C; [α] −19.0 (c 1.06, CHCl3)
(1S,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-carboxylic acid: (1S,2S)-6
Following the similar hydroxylation procedure described above, (1S,2S)-6 was prepared from (1S,2S)-4 (92%). White colored crystals; m.p. 101–104 °C; [α] +17.8 (c 1.00, CHCl3).
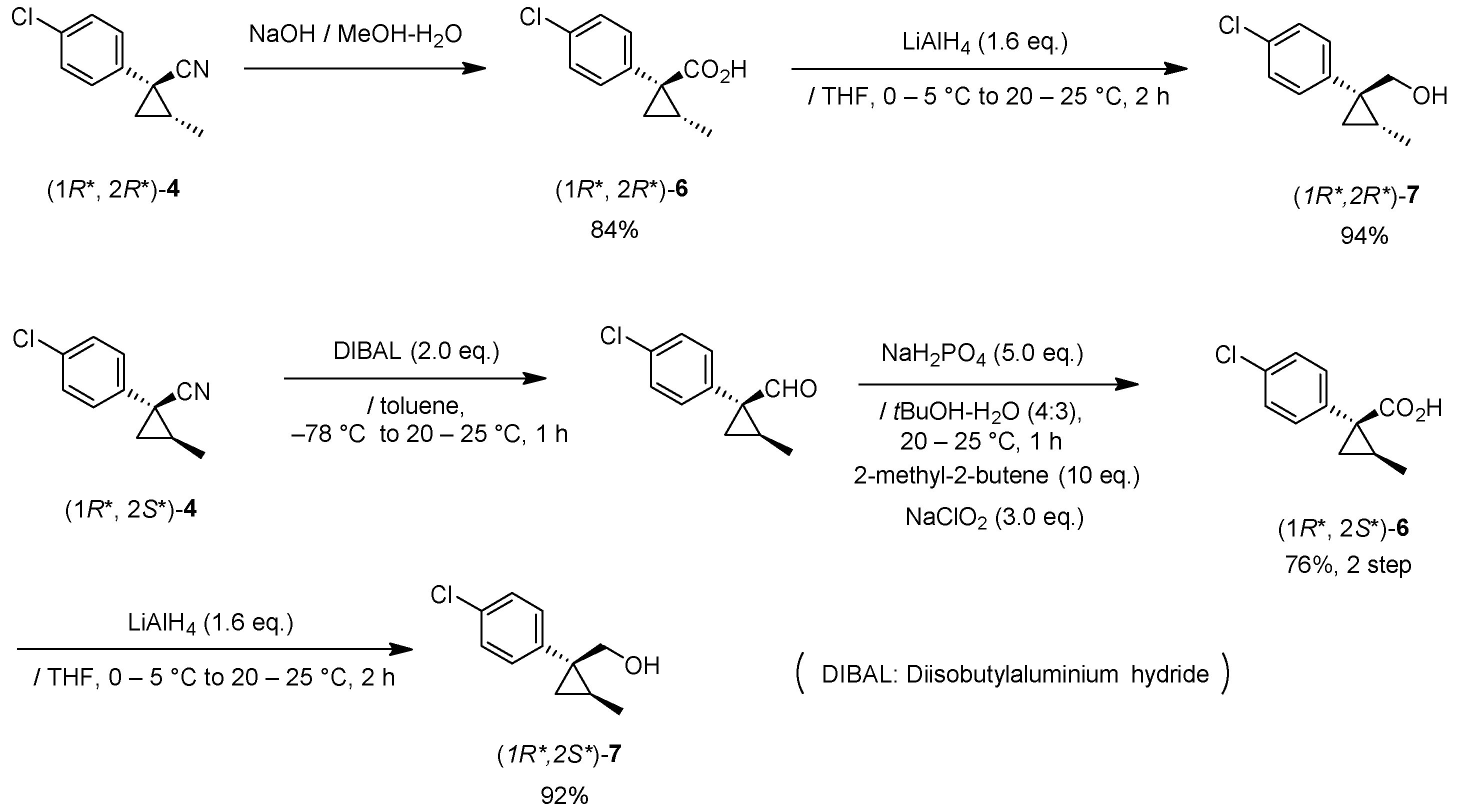
Cyano(3-phenoxyphenyl)methyl(1R*,2S*)-1-(4-chlorophenyl)-2-methylcyclopropane-1-carboxylate: (1R*,2S*)-8
![Molecules 24 01023 i016]()
Et3N (0.06 mL, 0.40 mmol) was added to a stirred mixture of (1R*,2S*)-6 (77 mg, 0.36 mmol) and bromo(3-phenoxyphenyl)acetonitrile (104 mg, 0.36 mmol) in acetone (0.72 mL) at 0–5 °C, and the mixture was stirred at 20–25 °C for 2 h. 1M HCl aq. solution was added to the mixture, which was extracted twice with ether. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel columm chromatography (hexane/EtOAc = 30:1) to give the desired product (1R*,2S*)-8 (110 mg, 73%). Yellow colored oil; 1H-NMR (400 MHz, CDCl3) δ = 1.26 (d, J = 6.0 Hz, 3H × 1/2), 1.32 (d, J = 6.0 Hz, 1H × 1/2), 1.32 (dd, J = 4.1 Hz, 4.6 Hz, 1H × 1/2), 1.36 (dd, J = 4.6 Hz, 8.7 Hz, 1H × 1/2), 1.58 (dd, J = 4.6 Hz, 5.0 Hz, 1H × 1/2), 1.60 (dd, J = 4.6 Hz, 5.0 Hz, 1H × 1/2), 1.63–1.73 (m, 1H), 6.31 (s, 1H × 1/2), 6.34 (s, 1H × 1/2), 6.87–7.49 (m, 13H); 13C-NMR (125 MHz) δ = 12.8, 12.9, 22.5, 22.7, 25.0, 25.2, 33.8, 33.9, 62.6, 62.7, 115.7, 116.0, 116.9, 117.0, 119.2, 119.4, 120.1, 120.2, 121.4, 121.6, 124.0, 124.1, 128.5 (2C), 123.0 (2C), 130.5, 130.6, 131.4 (2C), 133.3 (2C), 133.4, 133.6, 138.0 (2C), 156.2, 156.3, 158.0, 158.2, 170.2 (2C); νmax (neat)/cm−1 2931, 2360, 1734, 1585, 1487, 1155, 1244, 1090, 1015, 692. HRMS (DART) calcd for C25H20ClNO3 [M + H]+ 418.1210, found 418.1192.
Cyano(3-phenoxyphenyl)methyl (1R*,2R*)-1-(4-chlorophenyl)-2-methylcyclopropane-1-carboxylate: (1R*,2R*)-8
Following the similar esterification procedure described above, (1R*,2R*)-8 was prepared from (1R*,2R*)-6 (79%). Yellow colored oil; 1H-NMR (400 MHz, CDCl3) δ = 0.83 (d, J = 5.8 Hz, 3H × 1/2), 0.84 (d, J = 4.8 Hz, 3H × 1/2), 1.13 (dd, J = 4.5 Hz, 4.8 Hz, 1H × 1/2), 1.16 (dd, J = 4.1 Hz, 4.8 Hz, 1H × 1/2), 1.81 (dd, J = 4.5 Hz, 9.3 Hz, 1H × 1/2), 1.84 (dd, J = 4.5 Hz, 9.3 Hz, 1H × 1/2), 1.89–2.00 (m, 1H), 6.29 (s, 1H × 1/2), 6.32 (s, 1H × 1/2), 6.92–7.49 (m, 13H); 13C-NMR (125 MHz) δ = 15.2, 15.2, 23.6(2C), 24.1, 24.3, 32.7(2C), 62.6(2C), 115.8, 115.9, 116.8, 116.9, 119.3(2C), 119.3(2C), 120.0, 120.1, 121.4, 121.5, 124.0(2C), 128.4(2C), 129.9(2C), 130.5(2C), 132.7(2C), 133.1(2C), 133.4, 133.4, 133.5(2C), 156.2, 156.2, 158.1(2C), 172.2, 172.2; νmax (neat)/cm−1 2960, 2359, 1738, 1585, 1487, 1207, 1163, 1112, 1074, 1045, 745, 690.
(1R*,2S*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1R*,2S*)-7
![Molecules 24 01023 i018]()
Acid (1R*,2R*)-7 (126 mg, 0.6 mmol) in THF (3.0 mL) was added to a stirred suspension of LiAlH4 (36 mg) in THF (1.0 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at 20–25 °C for 2 h. Sat. NH4Cl aqueous solution was added to the mixture, which was filtered through Celite® using EtOAc. The separated organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/EtOAc = 10:1) to give the desired product (1R*,2R*)-7 (102 mg, 92%). Colorless oil; 1H-NMR (400 MHz) δ = 0.57 (dd, J = 5.0 Hz, 5.0 Hz, 1H), 1.01 (dd, J = 5.0 Hz, 8.2 Hz, 1H), 1.19–1.27 (m, 2H), 1.31 (d, J = 6.0 Hz, 3H), 3.74 (d, J = 11.9 Hz, 1H), 3.91 (d, J = 11.9 Hz, 1H), 7.21–7.36 (m, 4H); 13C-NMR (125 MHz) δ = 13.9, 19.0, 19.2, 31.2, 66.7, 128.4, 130.3, 132.1, 143.1; νmax (neat)/cm−1 3402, 2930, 1493, 1036, 1016, 831. HRMS (DART) calcd for C11H13ClO [M + H]+ 196.0655, found 196.0651.
(1R*,2R*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1R*,2R*)-7
Following the similar reduction procedure described above, (1R*,2R*)-7 was prepared from (1R*,2R*)-6 (94%). Colorless oil; 1H-NMR (400 MHz, CDCl3) δ = 0.61 (dd, J = 4.8 Hz, J = 4.8 Hz, 1H), 0.80 (d, J = 6.2 Hz, 3H), 0.97 (dd, J = 4.8 Hz, J = 8.7 Hz, 1H), 1.06–1.14 (m, 1H), 1.25-1.45 (s, 1H), 3.38 (d, J = 11.2 Hz, 1H), 3.77 (d, J = 11.2 Hz, 1H), 7.24–7.32 (m, 4H); 13C-NMR (125 MHz) δ = 15.4, 16.1, 16.7, 32.8, 71.7, 128.3, 132.0, 132.2, 138.3; νmax (neat)/cm−1 3347, 2868, 1492, 1091, 1015, 827, 737.
(1R,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1R,2S)-7
Following the similar reduction procedure described above, (1R,2S)-7 was prepared from (1R,2S)-6 (93%). Colorless oil; [α] +56.6 (c 1.12, CHCl3).
(1S,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1S,2R)-7
Following the similar reduction procedure described above, (1S,2R)-7 was prepared from (1S,2R)-6 (92%). Colorless oil; [α] −54.9 (c 0.46, CHCl3).
(1R,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1R,2R)-7
Following the similar reduction procedure described above, (1R,2R)-7 was prepared from (1R,2R)-6 (92%). White colored crystals; m.p. 56‒57 °C; [α] −31.9 (c 0.95, CHCl3).
(1S,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methanol: (1S,2S)-7
Following the similar reduction procedure described above, (1S,2S)-7 was prepared from (1S,2S)-6 (93%). White colored crystals; m.p. 51‒53 °C; [α] +32.4 (c 0.52, CHCl3).
(1R*,2S*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1R*,2S*)-9
![Molecules 24 01023 i024]()
A mixture of (1R*,2S*)-7 (183 mg, 0.93 mmol) and 3-phenoxybenzyl bromide (295 mg, 1.12 mmol) in DMF (0.9 mL) was added to a stirred suspension of NaH (60% dispersion; 45 mg, 1.12 mmol) at at 0–5 °C under an Ar atmosphere, and the mixture was stirred at 20–25 °C for 2 h. The mixture was quenched by water, which was extracted by EtOAc twice. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/EtOAc = 30:1) to give the desired product (1R*,2S*)-9 (322 mg, 91%). Colorless oil; 1H-NMR (400 MHz, CDCl3) δ = 0.53 (dd, J = 4.6 Hz, J = 4.6 Hz, 1H), 1.01 (dd, J = 4.6 Hz, J = 8.2 Hz, 1H), 1.14–1.28 (m, 4H), 3.51 (d, J = 10.1 Hz, 1H), 3.73 (d, J = 10.1 Hz, 1H), 4.39 (d, J = 12.4 Hz, 1H), 4.44 (d, J = 12.4 Hz, 1H), 6.90–7.01 (m, 5H), 7.10–7.27 (m, 6H), 7.32–7.36 (m, 2H); 13C-NMR (125 MHz) δ = 14.1, 19.5, 19.7, 28.8, 72.4, 73.9, 117.6, 117.8, 119.0, 122.1, 123.3, 128.1, 129.5, 129.7, 129.8, 131.5, 140.6, 143.9, 157.1, 157.4.; νmax (neat)/cm−1 2859, 1584, 1445, 1213, 1092, 1076, 957, 831, 748; HRMS (DART) calcd for C24H23ClO2 [M + H]+ 379.1465, found 379.1447.
(1R*,2R*)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1R*,2R*)-9
Following the similar etherification procedure described above, (1R*,2R*)-9 was synthesized from (1R*,2R*)-7 (79%). Colorless oil; 1H-NMR (400 MHz, CDCl3) δ = 0.62 (dd, J = 4.8 Hz, J = 4.8 Hz, 1H), 0.76 (d, J = 6.2 Hz, 3H), 0.94 (dd, J = 4.8 Hz, J = 8.7 Hz, 1H,), 1.03–1.11 (m, 1H), 3.33 (d, J = 9.9 Hz, 1H), 3.53 (d, J = 9.9 Hz, 1H), 4.38 (dd, J = 12.4 Hz, J = 14.7 Hz, 2H), 6.86–6.91 (m, 3H), 6.98–7.01 (m, 2H), 7.09–7.14 (m, 1H), 7.22–7.24 (m, 5H), 7.31–7.37 (m, 2H); 13C-NMR (125 MHz) δ = 15.4, 16.3, 17.0, 30.5, 72.1, 78.9, 117.4, 117.7, 118.9, 121.9, 123.2, 128.0, 129.5, 129.7, 131.9, 132.0, 138.9, 140.6, 157.0, 157.3; νmax (neat)/cm−1 2855, 1584, 1445, 1250, 1213, 1092, 1015, 743.
(1R,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1R,2S)-9
Following the similar etherification procedure described above, (1R,2S)-9 was synthesized from (1R,2S)-7 (85%). Colorless oil; [α] +25.8 (c 1.19, CHCl3).
(1S,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1S,2R)-9
Following the similar etherification procedure described above, (1S,2R)-9 was synthesized from (1S,2R)-7 (72%). Colorless oil; [α] ‒24.5 (c 0.50, CHCl3).
(1R,2R)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1R,2R)-9
Following the similar etherification procedure described above, (1R,2R)-9 was synthesized from (1R,2R)-7 (91%). Colorless oil; [α] −31.8 (c 0.84, CHCl3).
(1S,2S)-1-(4-Chlorophenyl)-2-methylcyclopropane-1-methyl 3-phenoxybenzyl ether: (1S,2S)-9
Following the similar etherification procedure described above, (1S,2S)-9 was synthesized from (1S,2S)-7 (97%). Colorless oil; [α] +32.4 (c 0.65, CHCl3).
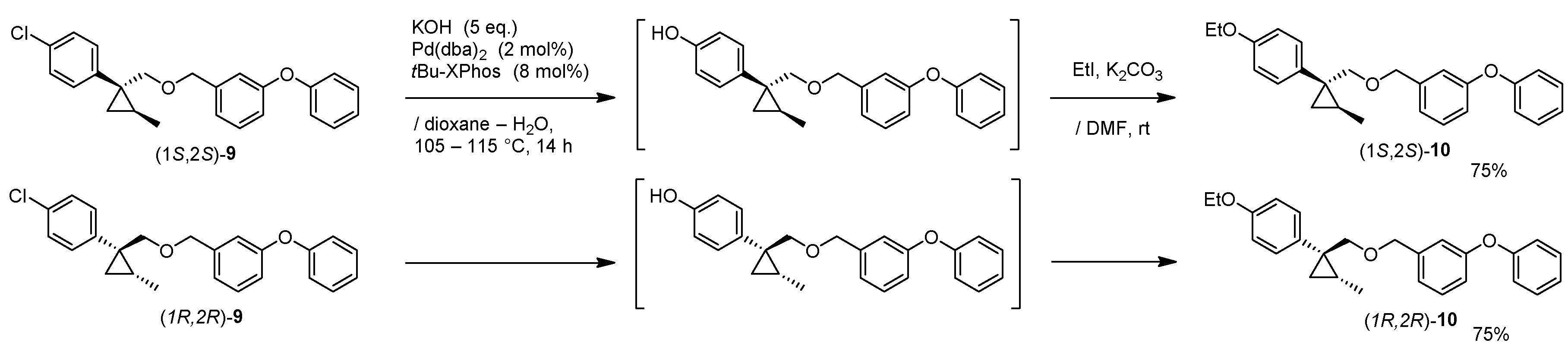
1-((((1R,2R)-1-(4-Ethoxyphenyl)-2-methylcyclopropyl)methoxy)methyl)-3-phenoxybenzene: (1R,2R)-10
A mixture of (1R,2R)-9 (108 mg, 0.29 mmol), Pd2(dba)3 (5.3 mg, 2 mol%), tBu-XPhos (10 mg, 8 mol%) and KOH (81 mg, 5.0 eq.) in 1,4-dioxane (0.3 mL) and H2O (0.3 mL) was stirred at 100–105 °C for 16 h under an Ar atmosphere. The mixture was cooled down to room temperature, and quenched with water, which was extracted twice with EtOAc. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/EtOAc = 20:1) to give the desired phenol product (95 mg, 90%).
K2CO3 (43 mg, 0.31 mmol) and EtI (122 mg, 0.78 mmol) was successively added to a stirred solution of phenol (92 mg, 0.26 mmol) in DMF (0.5 mL) at 0–5 °C under an Ar atmosphere. The mixture was stirred at 20–25 °C for 16 h. The mixture was quenched with water, which was extracted three times with EtOAc. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude oil was purified by SiO2-gel column chromatography (hexane/EtOAc = 20:1) to give the desired product (1R,2R)-10 (84 mg, 83%).
(1R,2R)-10: Colorless oil; [α] −23.4 (c 0.95, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 0.60 (dd, J = 4.8 Hz, J = 4.8 Hz, 1H), 0.77 (d, J = 6.2 Hz, 3H), 0.90 (dd, J = 4.8 Hz, J = 8.2 Hz, 1H), 0.99–1.07 (m, 1H), 1.40 (t, J = 7.1 Hz, 3H), 3.34 (d, J = 9.9 Hz, 1H), 3.54 (d, J = 9.9 Hz, 1H), 4.00 (q, J = 7.1 Hz, 2H), 4.39 (t, J = 13.3 Hz, 2H), 6.78–6.82 (m, 2H), 6.86–6.88 (m, 2H), 6.93 (d, J = 8.0 Hz, 1H), 6.98–7.00 (m, 2H), 7.08–7.12 (m, 1H), 7.19–7.22 (m, 2H), 7.23–7.25 (m, 1H), 7.31–7.35 (m, 2H); 13C-NMR (125 MHz) δ = 14.9, 15.4, 16.2, 17.0, 30.2, 63.2, 72.1, 79.3, 113.9, 117.6, 117.7, 118.9, 122.0, 123.2, 129.5, 129.7, 131.5, 132.2, 140.9, 157.2, 157.2, 157.4; νmax (neat)/cm−1 2980, 2868, 1611, 1584, 1514, 1487, 1445, 1393, 1354, 1288, 1242, 1215, 1175, 1140, 1107, 1074, 1049, 959, 924, 835, 789, 735, 692; HRMS (ESI): m/z calcd for C26H28O3 [M + Na]+ 411.1936; found: 411.1936.
(1R,2R)-10: Pale yellow oil; [α] +24.7 (c 0.21, CHCl3).












































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












