Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion


Abstract
:
1. Introduction
2. Results and Discussion
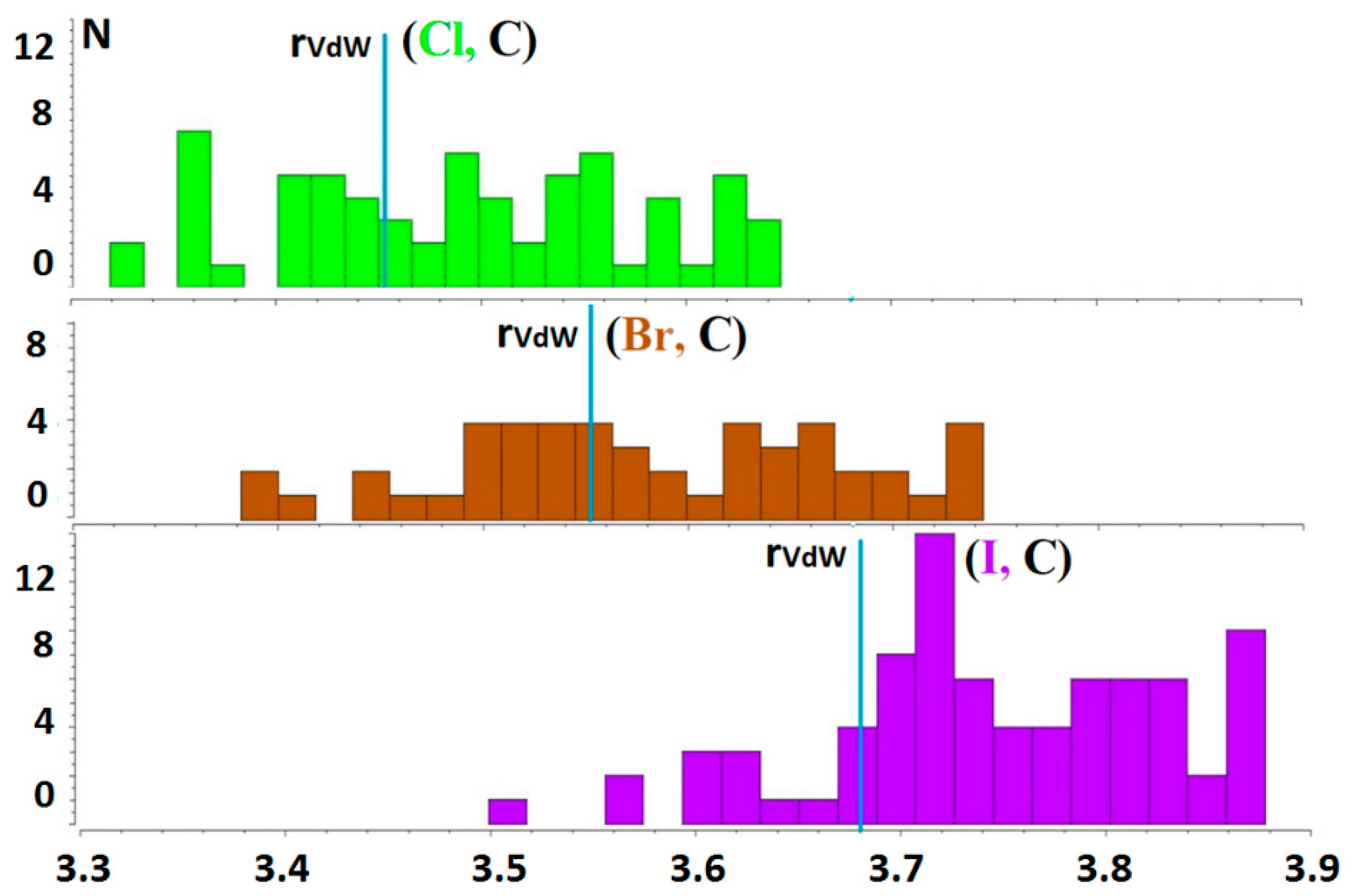
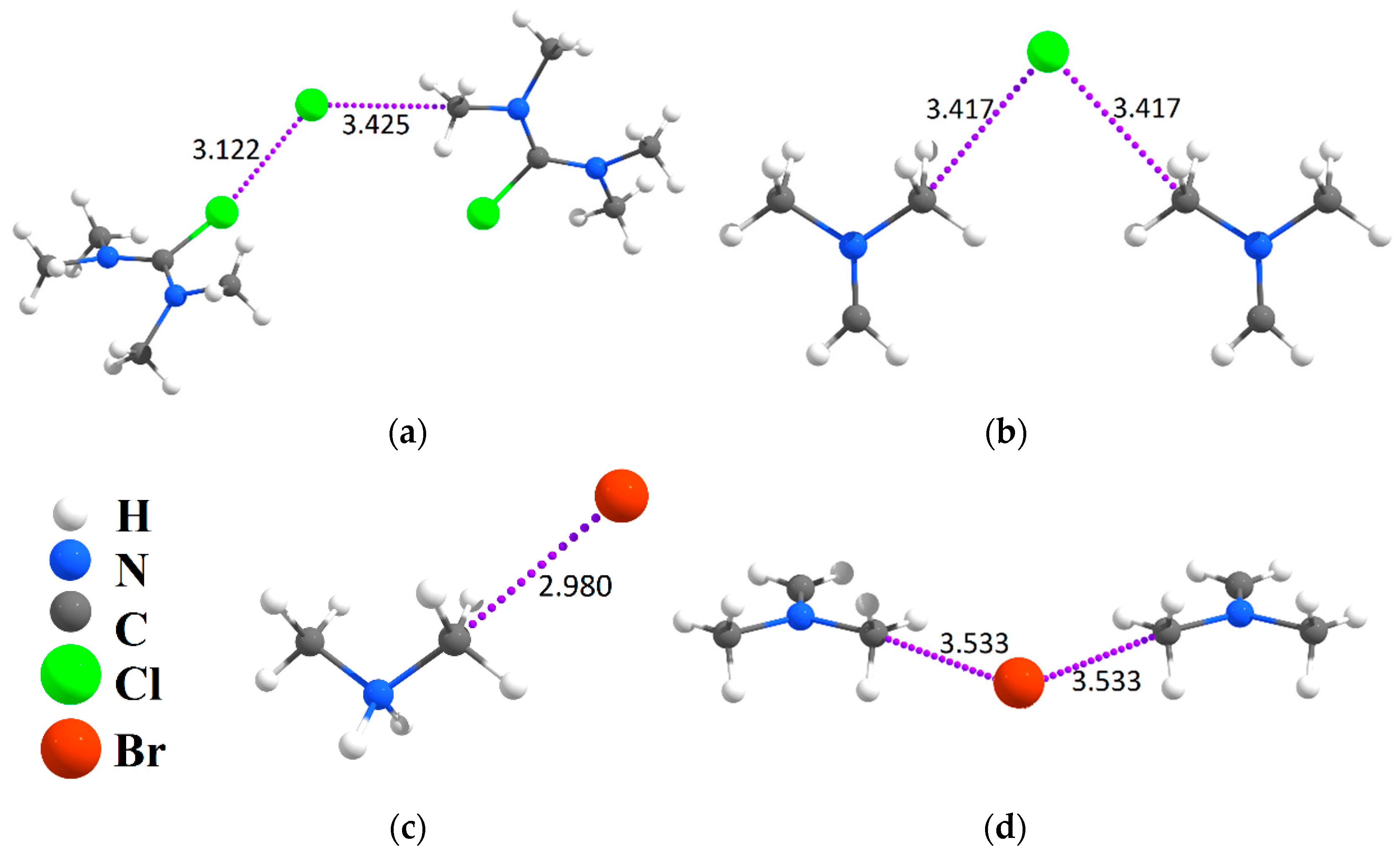
2.1. Population of Hal−···CH3Y Tetrel Bonds in Crystals
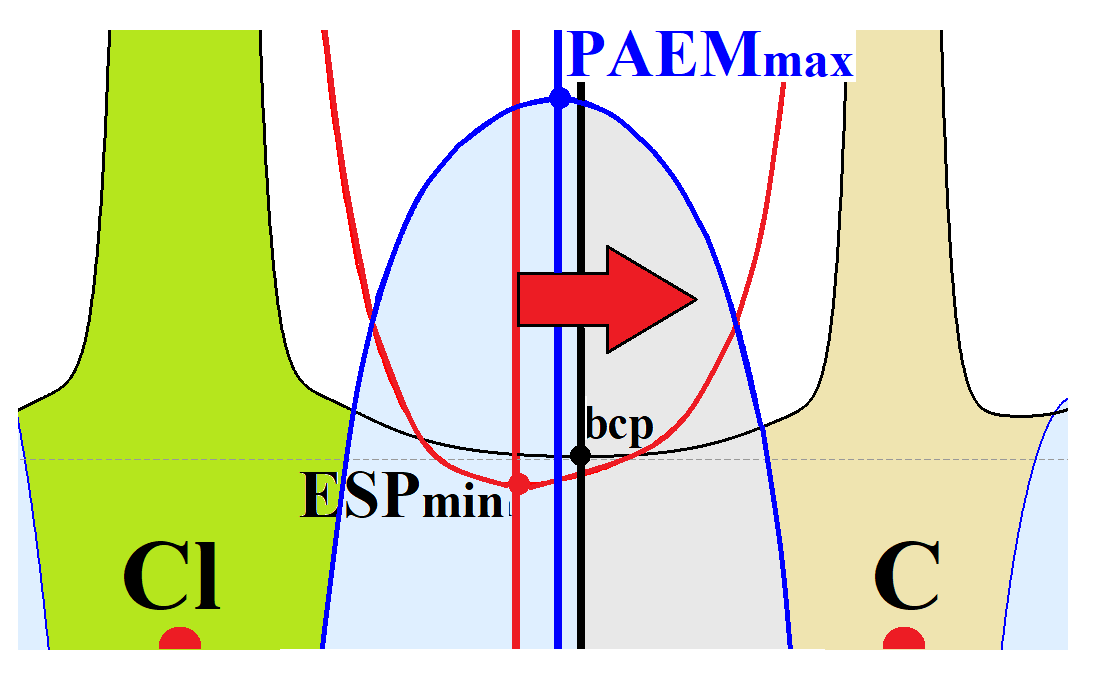
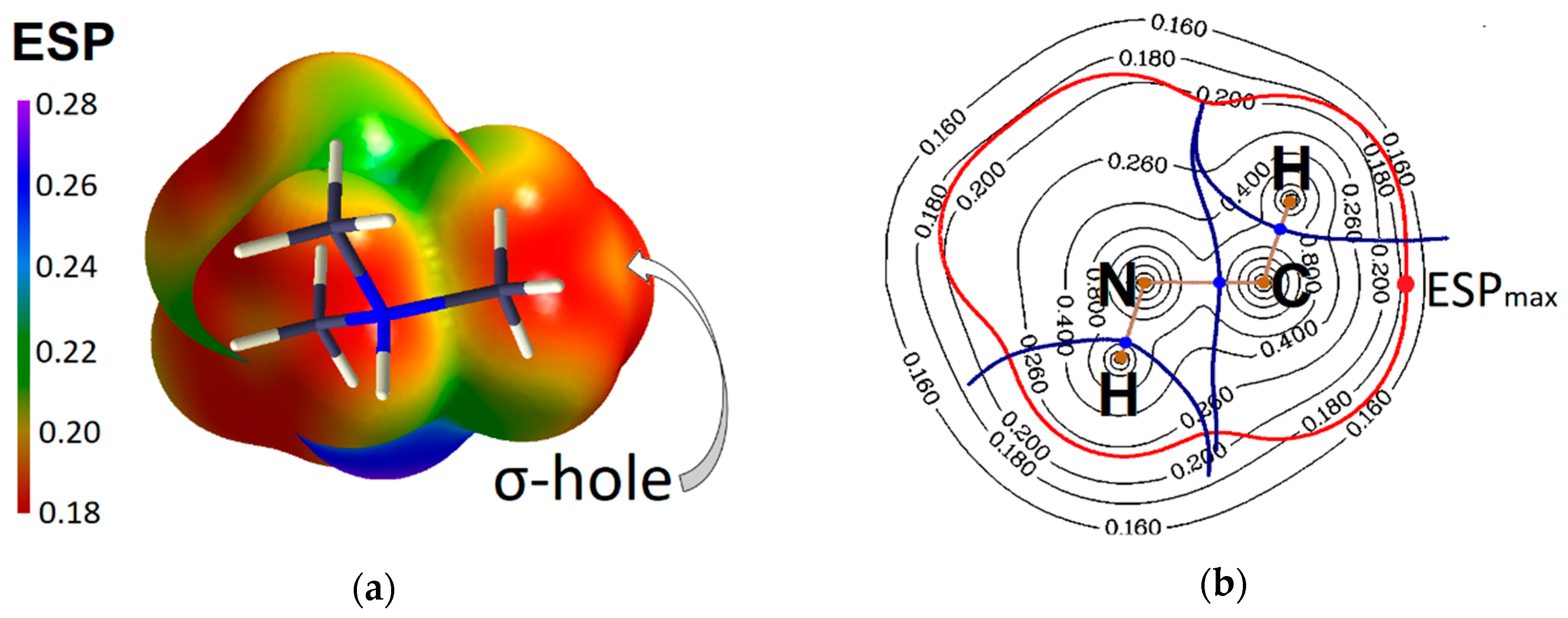
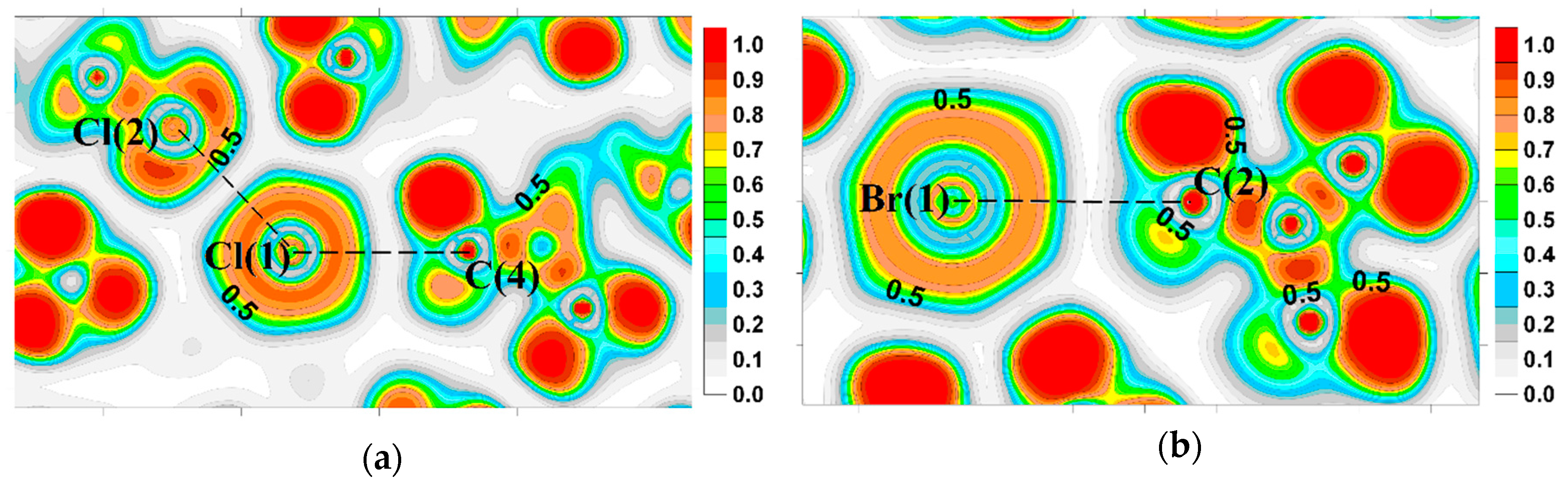
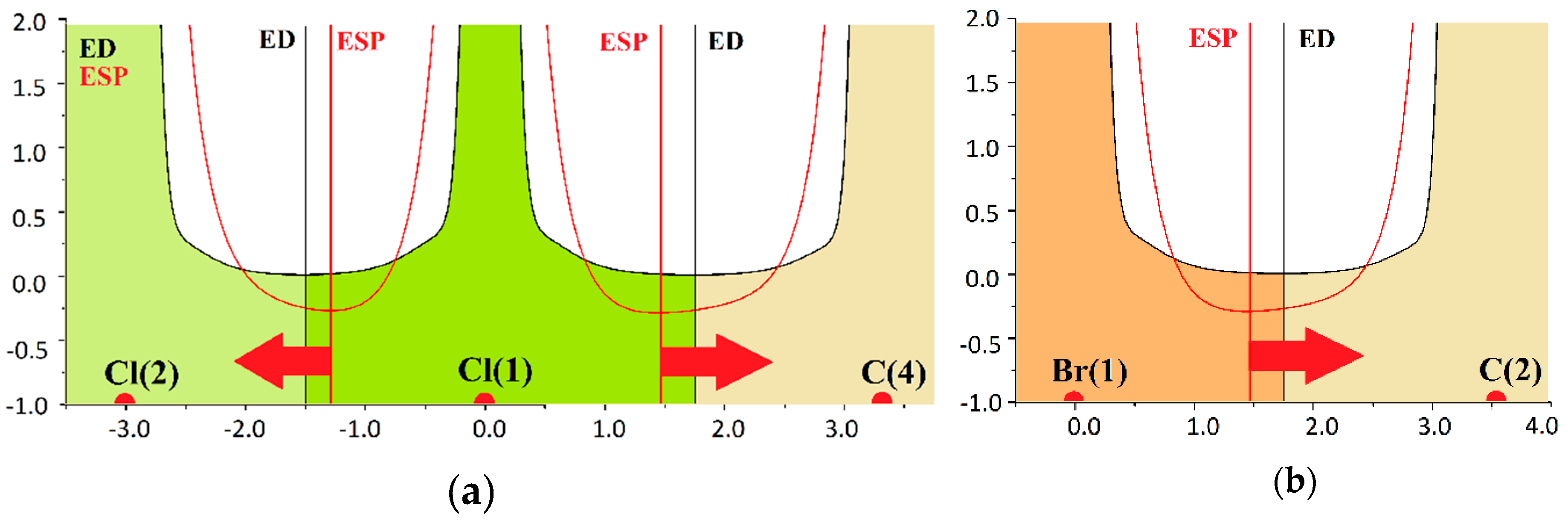
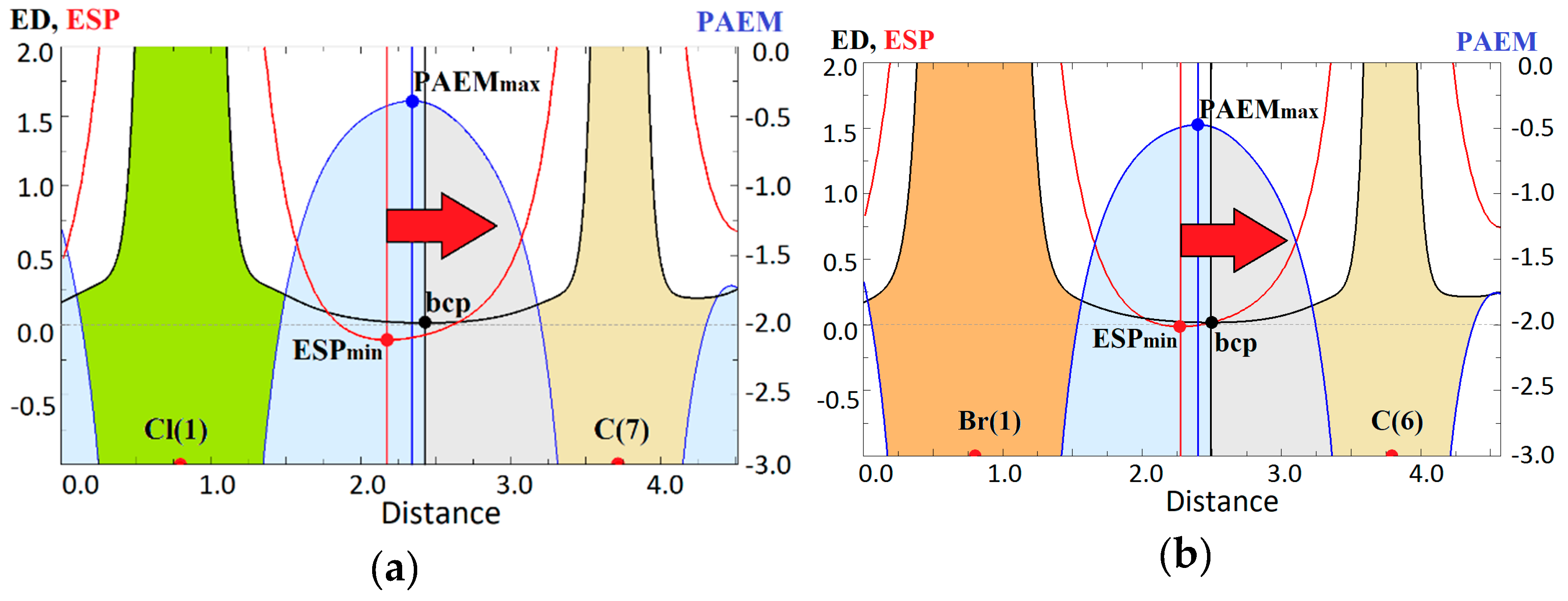
2.2. Evidence of Electrophilic Sites for the CH3-Groups Bound in Tetrel Bonds
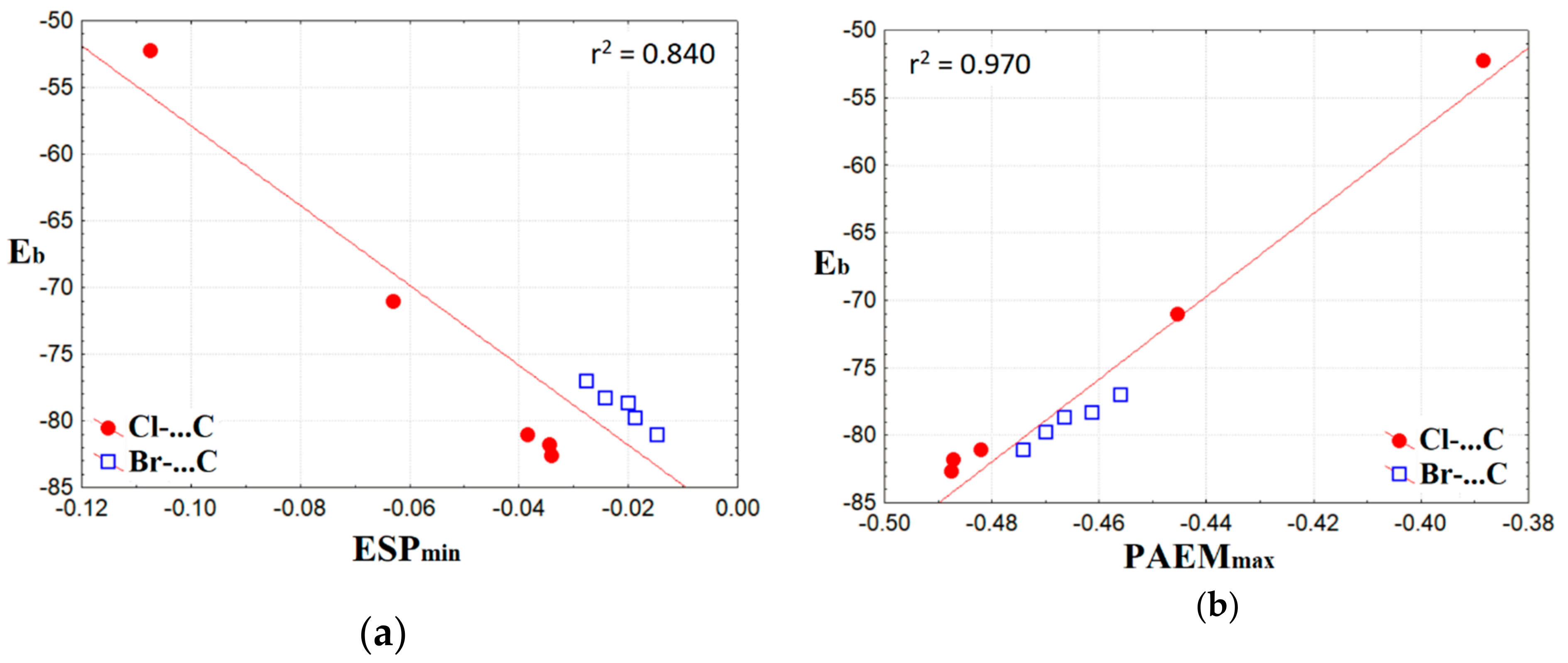
2.3. Binding Energy in Molecular Complexes with the Hal−···CH3 Tetrel Bonds
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Legon, A.C. Tetrel, Pnictogen and Chalcogen Bonds Identified in the Gas Phase before they had Names: A Systematic Look at Non-covalent Interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The Nature of Halogen···Halogen Interactions: Are Short Halogen Contacts Due to Specific Attractive Forces or Due to Close Packing of Nonspherical Atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole Revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Anionic Complexes of F− and Cl− with Substituted Methanes: Hydrogen, Halogen, and Tetrel Bonds. Chem. Phys. Lett. 2016, 655–656, 115–119. [Google Scholar] [CrossRef]
- Scheiner, S. Tetrel Bonding as a Vehicle for Strong and Selective Anion Binding. Molecules 2018, 23, 1147. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S.; Mousavian, P. Anionic Tetrel Bonds: An ab Initio Study. Chem. Phys. Lett. 2018, 691, 394–400. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. Strong Tetrel Bonds: Theoretical Aspects and Experimental Evidence. Molecules 2018, 23, 2642. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C···Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A. RCH3···O Interactions in Biological Systems: Are They Trifurcated H-Bonds or Noncovalent Carbon Bonds? Crystals 2016, 6, 26. [Google Scholar] [CrossRef]
- Garcia-LLinas, X.; Bauza, A.; Seth, S.K.; Frontera, A. Importance of R−CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Konar, S.; Lama, P.; Das, K.; Bauza, A.; Frontera, A.; Mukhopadhyay, S. On the Importance of Noncovalent Carbon Bonding Interactions in the Stabilization of a 1D Co(II) Polymeric Chain as Precursor of a Novel 2D Coordination Polymer. J. Phys. Chem. B 2016, 120, 6803–6811. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Stash, A.I.; Potemkin, V.A.; Rykounov, A.A.; Shutalev, A.D.; Zhurova, E.A.; Zhurov, V.V.; Pinkerton, A.A.; Gurskaya, G.V.; Zavodnik, V.E. Molecular and Crystal Properties of Ethyl-4,6-dimethyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate from Experimental and Theoretical Electron Densities. Acta Cryst. B 2006, B62, 676–688. [Google Scholar] [CrossRef]
- Thomas, S.P.; Pavan, M.S.; Row, T.N.G. Experimental Evidence for ‘Carbon Bonding’ in the Solid State from Charge Density Analysis. Chem. Commun. 2014, 50, 49–51. [Google Scholar] [CrossRef]
- Scheiner, S. Steric Crowding in Tetrel Bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk1, M.; Scheiner, S. Comparison between Tetrel Bonded Complexes Stabilized by σ and π Hole Interactions. Molecules 2018, 23, 1416. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic Elucidation of Factors That Influence the Strength of Tetrel Bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of Tetrel Bonds in Neutral and Protonated Complexes of PyridineTF3 and FuranTF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Laconsay, C.J.; Galbraith, J.M. A Valence Bond Theory Treatment of Tetrel Bonding Interactions. Comp. Theor. Chem. 2017, 1116, 202–206. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk1, M.; Scheiner, S. Implications of Monomer Deformation for Tetrel and Pnicogen Bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Stash, A.I.; Chen, Y.S.; Kovalchukova, O.V.; Tsirelson, V.G. Electron Density, Electrostatic Potential, and Spatial Organization of Ammonium Hydrooxalate Oxalic Acid Dihydrate Heteromolecular Crystal from Data of Diffraction Experiment at 15 K Using Synchrotron Radiation and Theoretical Calculations. Russ. Chem. Bull. 2013, 62, 1752–1763. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; pp. 1–438. [Google Scholar]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Avilov, A.S.; Lepeshov, G.G.; Kulygin, A.K.; Stahn, J.; Pietsch, U.; Spence, J.C.H. Quantitative Analysis of the Electrostatic Potential in Rock-Salt Crystals Using Accurate Electron Diffraction Data. J. Phys. Chem. 2001, B105, 5068–5074. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Shishkina, A.V.; Stash, A.I.; Parsons, S. The Experimental and Theoretical QTAIMC Study of the Atomic and Molecular Interactions in Dinitrogen Tetroxide. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2009, B65, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Topological Properties of the Electrostatic Potential in Weak and Moderate N···H Hydrogen Bonds. J. Phys. Chem. 2007, A111, 6425–6433. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Yushina, I.D.; Kropotina, K.K.; Muhitdinova, S.E.; Tsirelson, V.G. Testing the Tools for Revealing and Characterizing the Iodine-Iodine Halogen Bond in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, B73, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Gadre, S.R. Maximal and Minimal Characteristics of Molecular Electrostatic Potentials. J. Chem. Phys. 1990, 93, 1770–1773. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Yushina, I.D.; Stash, A.I.; Tsirelson, V.G. Halogen Bonding and Other Iodine Interactions in Crystals of Dihydrothiazolo(oxazino)quinolinium Oligoiodides from the Electron-Density Viewpoint. Cryst. Growth Des. 2014, 14, 5674–5684. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Yushina, I.D.; Muhitdinova, S.E.; Tsirelson, V.G. Electronic Criterion for Categorizing the Chalcogen and Halogen Bonds: Sulfur—Iodine Interactions in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, in press. [Google Scholar] [CrossRef]
- Zhao, D.-X.; Gong, L.-D.; Yang, Z.-Z. The Relations of Bond Length and Force Constant with the Potential Acting on an Electron in a Molecule. J. Phys. Chem. A 2005, 109, 10121–10128. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.V.; Tsirelson, V.G. A Comparative View on the Potential Acting on an Electron in a Molecule and the Electrostatic Potential through the Typical Halogen Bonds. J. Comput. Chem. 2018, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.V.; Mukhitdinova, S.E.; Tsirelson, V.G. Characterizing the Halogen and Chalcogen Bonds in Crystals: PAEM vs ESP. In Book of Abstracts of International Union of Crystallography (IUCr)’s Sagamore XIX Conference on Quantum Crystallography, Halifax, NS, Canada; Mount Saint Vincent University’s Printshop: Halifax, Canada, 2018; pp. 93–95. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Tiritiris, I.; Kantlehner, W. Crystal Structure of N,N,N′,N′-tetramethylchloroformamidinium chloride, [C5H12N2Cl]Cl. Z. Kristallogr. New Cryst. Struct. 2008, 223, 345–346. [Google Scholar] [CrossRef]
- Burg, A.B. Restudy of the Action of Sulfur Dioxide on Dry Trimethylamine Oxide: Iodine Oxidation and Lewis Acid Chemistry of the Most Reactive Product, (CH3)2(H)NCH2SO3. Inorg. Chem. 1989, 28, 1295–1300. [Google Scholar] [CrossRef]
- Clark, G.R.; Shaw, G.L.; Surman, P.W.J.; Taylor, M.J.; Steele, D. Preparation, Structure and Vibrational Spectrum of the Dimethylmethyleniminium Ion, Including the Role of Cationic Polymers in its Formation. J. Chem. Soc. Faraday Trans. 1994, 90, 3139–3144. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between Non-covalent Interactions in Complexes and Crystals with Halogen Bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Can Halogen Bond Energy be Reliably Estimated from Electron Density Properties at Bond Critical Point? The Case of the (A)nZ–Y···X− (X, Y = F, Cl, Br) Interactions. Int. J. Quantum Chem. 2018. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Wilson, A.K.; Woon, D.E.; Peterson, K.A.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. IX. The Atoms Gallium through Krypton. J. Chem. Phys. 1999, 110, 7667–7676. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theoret. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL14 User’s Manual; University of Torino: Torino, Italy, 2014. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Semi-empirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Gatti, C.; Casassa, S. Topond14 User’s Manual; University of Torino: Torino, Italy, 2014. [Google Scholar]
- Keith, T.A. AIMALL, Version 12.06.03, 2012 Professional. Available online: http://aim.tkgristmill.com (accessed on 20 February 2019).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kostenetskiy, P.; Semenikhina, P. SUSU Supercomputer Resources for Industry and Fundamental Science. In Proceedings of the Global Smart Industry Conference (GloSIC), Chelyabinsk, Russia, 13–15 November 2018; pp. 1–7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | Bond | Crystal Dexp, θ (Hal−···C–N)exp | Crystal Dcalc, θ (Hal−···C–N)calc | ρ(rbcp), Crystal |
|---|---|---|---|---|
| GETQIF | Сl(3)− ···C(2) | 3.4584 169.08 | 3.4260 163.91 | 0.0056 |
| C(2)–N(1) | 1.4815 | 1.4958 | 0.2441 | |
| LONGEB | Сl(1)− ···C(4) | 3.4251 175.28 | 3.4087 166.64 | 0.0068 |
| C(4)–N(2) | 1.4722 | 1.4740 | 0.2458 | |
| VAPREJ | Сl(1)− ···C(1) | 3.417 164.88 | 3.4385 164.49 | 0.0064 |
| C(1)–N(1) | 1.466 | 1.4747 | 0.2480 | |
| TMHYZC | Сl(1)− ···C(2) | 3.4374 174.96 | 3.4280 176.34 | 0.0062 |
| C(2) ···N(1) | 1.4976 | 1.5080 | 0.2406 | |
| ZENJAD | Сl(1)− ···C(7) | 3.5111 170.58 | 3.4644 171.72 | 0.0056 |
| C(7)–O(2) | 1.4471 | 1.4468 | 0.2306 | |
| LILLOH | Br(1)− ···C(2) | 3.533 167.25 | 3.5664 166.86 | 0.0061 |
| C(2)–N(1) | 1.474 | 1.4735 | 0.2490 | |
| FADXIR | Br(1)− ···C(6) | 3.6014 170.87 | 3.5722 173.15 | 0.0058 |
| C(6)–N(1) | 1.5025 | 1.5082 | 0.2371 | |
| POSTUM02 | Br(1)− ···C(1) | 3.7012 175.21 | 3.6667 174.15 | 0.0048 |
| C(1)–N(1) | 1.4852 | 1.4928 | 0.2432 | |
| ZZZGVM01 | Br(1)− ···C(2) | 3.742 168.65 | 3.7283 169.04 | 0.0042 |
| C(2)–N(1) | 1.474 | 1.4968 | 0.2437 | |
| ZZZUQO03 | Br(1)− ···C(1) | 3.685 171.12 | 3.6819 171.11 | 0.0049 |
| C(1)–N | 1.487 | 1.5039 | 0.2411 |
| Refcode | Tetrel Bond | Eb | Dcalc | ρ(rbcp) | ESPmin | PAEMmax |
|---|---|---|---|---|---|---|
| GETQIF | Сl−···CH3Y | −81.07 | 2.8262 | 0.019 | −0.038 | −0.4819 |
| LONGEB | Сl−···CH3Y | −71.05 | 2.8782 | 0.017 | −0.063 | −0.4454 |
| TMHYZC | Сl−···CH3Y | −82.67 | 2.8248 | 0.019 | −0.034 | −0.4874 |
| VAPREJ | Сl−···CH3Y | −81.80 | 2.8226 | 0.019 | −0.034 | −0.4870 |
| ZENJAD | Сl−···CH3Y | −52.28 | 2.9268 | 0.015 | −0.107 | −0.3883 |
| FADXIR | Br−···CH3Y | −81.09 | 2.9855 | 0.017 | −0.015 | −0.4741 |
| LILLOH | Br−···CH3Y | −78.67 | 2.9896 | 0.016 | −0.020 | −0.4664 |
| POSTUM02 | Br−···CH3Y | −79.82 | 2.9802 | 0.017 | −0.019 | −0.4699 |
| ZZZGVM01 | Br−···CH3Y | −77.97 | 2.9949 | 0.0165 | −0.024 | −0.4612 |
| ZZZUQO03 | Br−···CH3Y | −76.63 | 3.0015 | 0.0162 | −0.028 | −0.4560 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartashevich, E.; Matveychuk, Y.; Tsirelson, V. Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules 2019, 24, 1083. https://doi.org/10.3390/molecules24061083
Bartashevich E, Matveychuk Y, Tsirelson V. Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules. 2019; 24(6):1083. https://doi.org/10.3390/molecules24061083
Chicago/Turabian StyleBartashevich, Ekaterina, Yury Matveychuk, and Vladimir Tsirelson. 2019. "Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion" Molecules 24, no. 6: 1083. https://doi.org/10.3390/molecules24061083
APA StyleBartashevich, E., Matveychuk, Y., & Tsirelson, V. (2019). Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules, 24(6), 1083. https://doi.org/10.3390/molecules24061083