
Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study


Abstract
:1. Introduction
2. Results
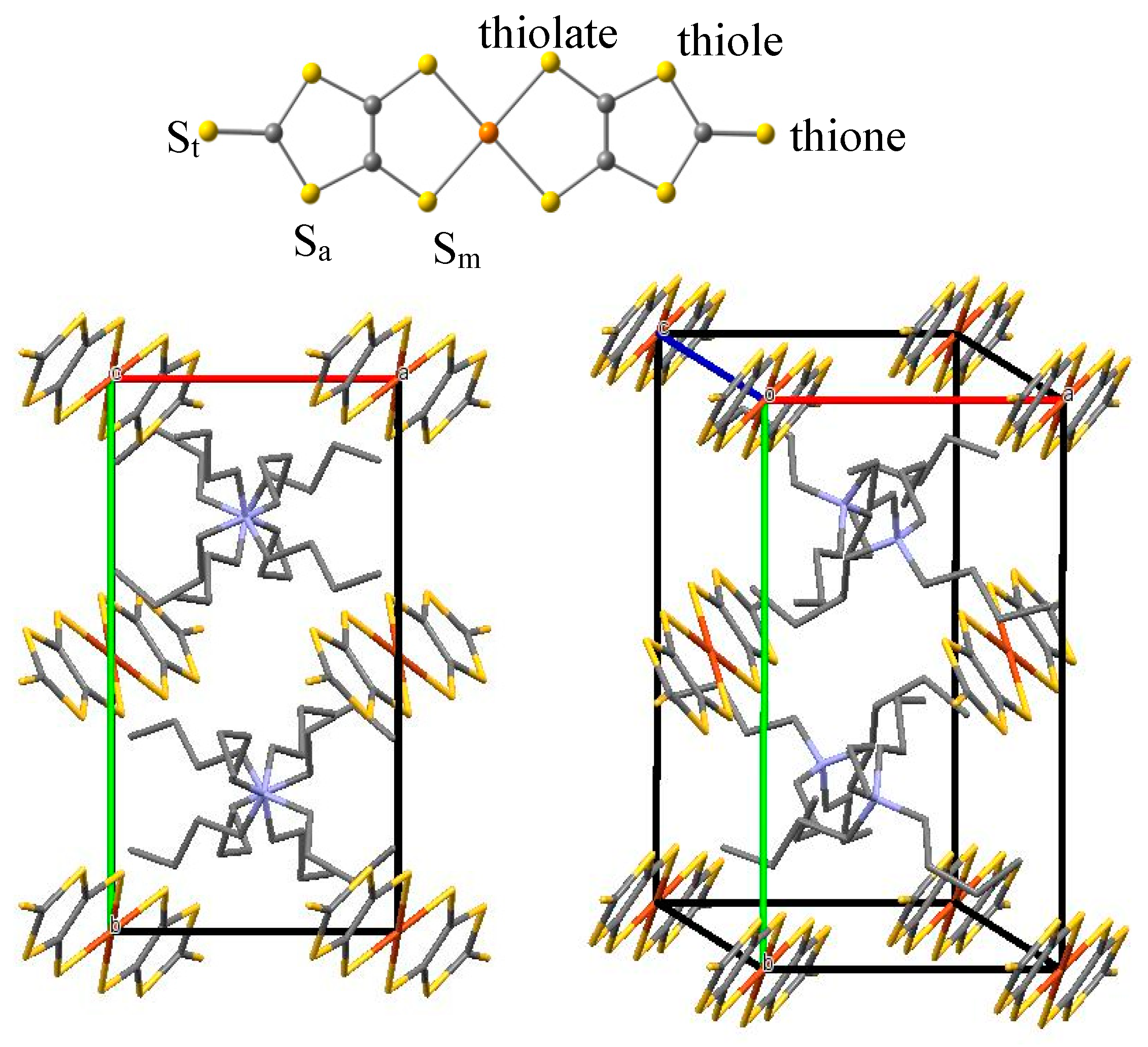
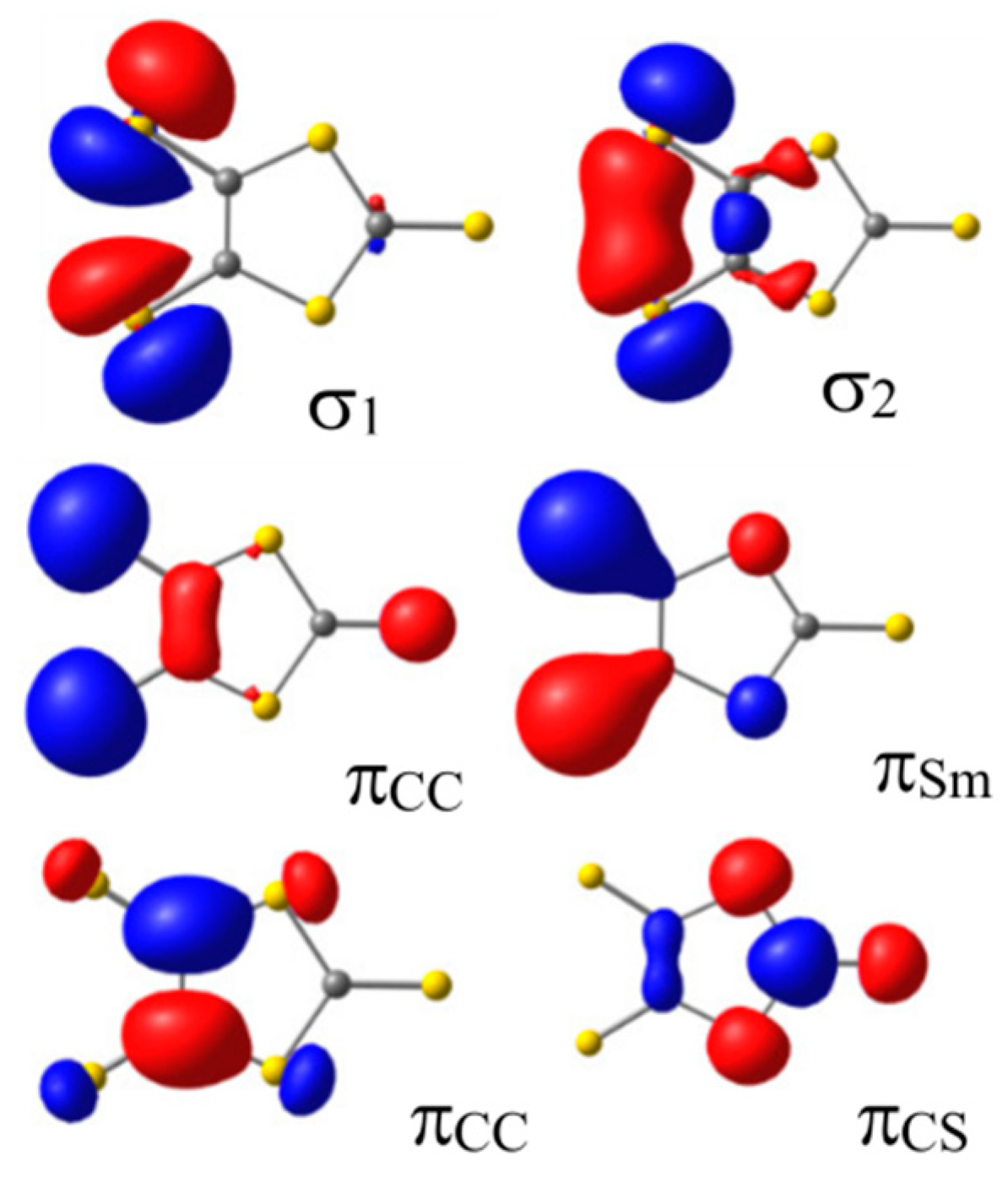
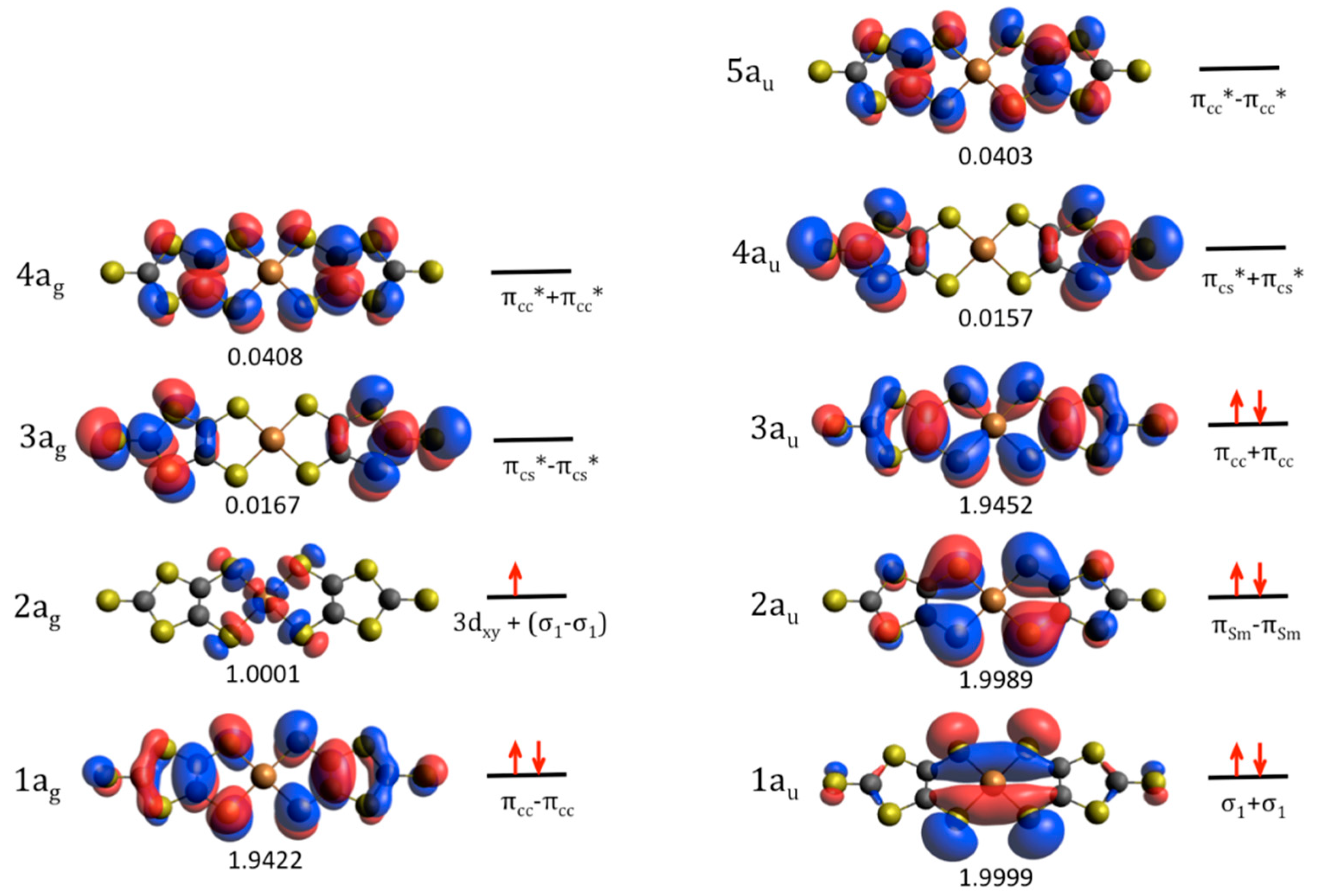
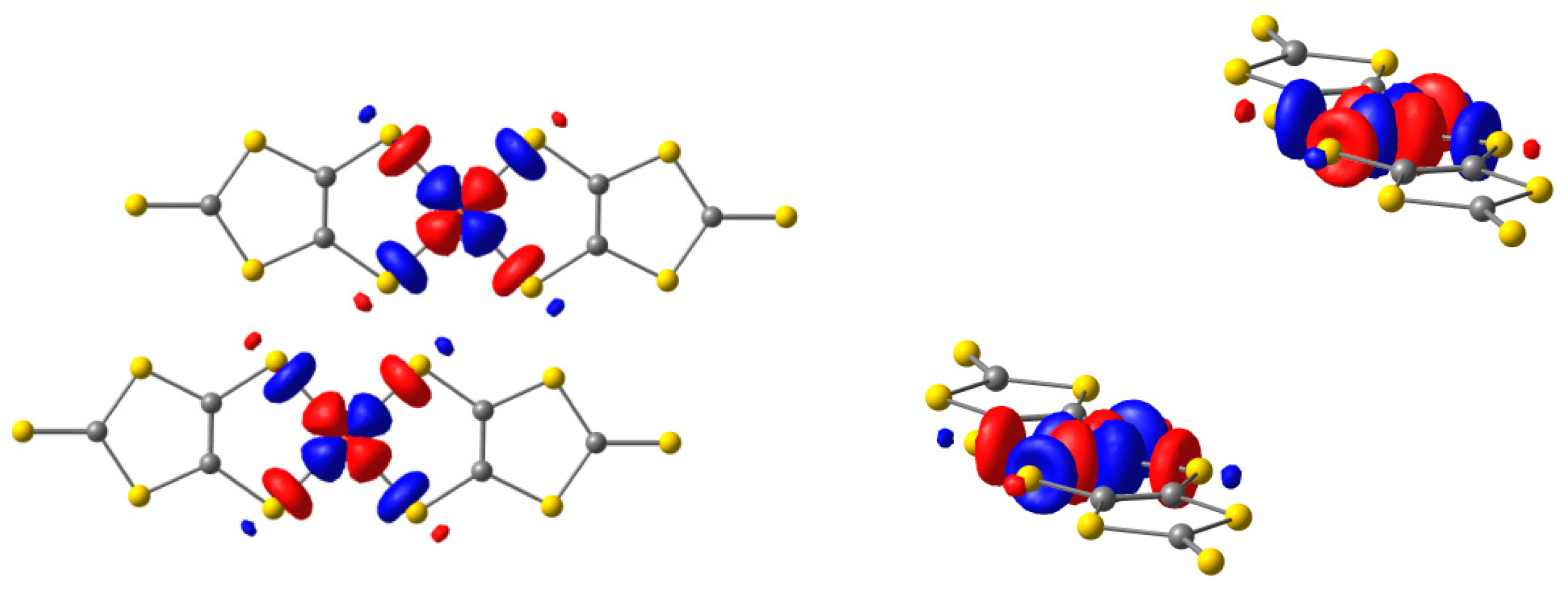
2.1. Ground State Wave Function of the Cu(dmit)2 Complex
2.2. Excited States
2.3. Magnetic Interactions
3. Discussion and Conclusions
- The excited states in the range of 2.0 to 5.5 eV present a noticeable multideterminantal character; the dominant configuration represents in most of the states no more than 50% of the whole wave function. This is a consequence of the presence of numerous virtual dmit π orbitals very close in energy, in such a way that the population of these empty orbitals by excitations from the occupied orbitals requires almost the same input of energy. As a result, in most of the explored states the spin density moves from the Cu 3dxy to the dmit π orbitals. This multideterminantal character makes difficult a proper description by means of single-reference methods such as DFT, TD-DFT and, of course, extended Hückel calculations, and could be in the origin of the discrepancies found between our analysis and those previously reported.
- Among these states, five of them present non-negligible oscillator strength, and then the excitations from the ground state are allowed by the electric-dipole selection rules. In these states, the spin density shows a marked change with respect to the ground state, i.e., it is mainly (or completely) localized on the dmit ligands. This feature agrees with the fact that the observed g-values (~2.00) under UV irradiation are comparable with that of the free electron, then in better agreement with the unpaired electrons placed on the organic dmit ligands than localized on the Cu centers.
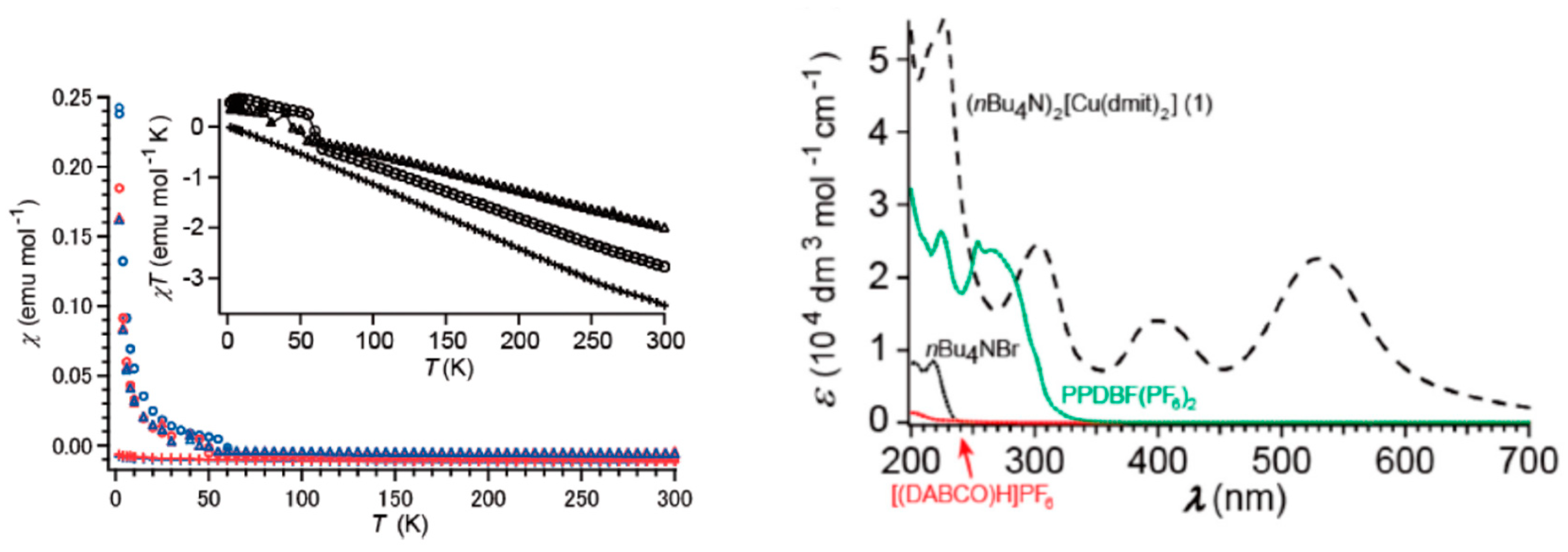
- The magnetic interactions between the [Cu(dmit)2]−2 complexes are very weak, slightly ferromagnetic, and in good agreement with both the long intermolecular distances, and the relative orientation of the SOMO orbitals in two close [Cu(dmit)2]−2 complexes. The experimental thermal dependence of the magnetic susceptibility data at low temperature (T < 50 K) can be simulated assuming isolated S = 1/2 units interacting with a very weak ferromagnetic coupling, by a Curie–Weiss law or a 1D-ferromagnetic chain spin model. The reported diamagnetic behavior found for T > 50 K for these salts is by no means due to a strong antiferromagnetic interaction between the [Cu(dmit)2]−2 units, as claimed in previous works [23,24]. Neither the J coupling value nor the crystal structure supports such a strong interaction.
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Day, P.; Coronado, E. Molecular materials combining magnetic and conducting properties. In Magnetism: Molecules to Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar]
- Fourmigué, M.; Ouahab, L. Conducting and Magnetic Organometallic Molecular Materials; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Robertson, N.; Cronin, L. Metal bis-1,2-dithiolene complexes in conducting or magnetic crystalline assemblies. Coord. Chem. Rev. 2002, 227, 93–127. [Google Scholar] [CrossRef]
- Kobayashi, H.; Miyamoto, A.; Kato, R.; Sakai, F.; Kobayashi, A.; Yamakita, Y.; Furukawa, Y.; Tasumi, M.; Watanabe, T. Mixed valency of Cu, electron-mass enhancement, and three-dimensional arrangement of magnetic sites in the organic conductors (R1,R2-N,N’-dicyanoquinonediimine)2Cu (where R1,R2=CH3,CH3O,Cl,Br). Phys. Rev. B 1993, 47, 3500–3510. [Google Scholar] [CrossRef]
- Uji, S.; Terashima, T.; Aoki, H.; Brooks, J.S.; Kato, R.; Sawa, H.; Aonuma, S.; Tamura, M.; Kinoshita, M. Coexistence of one- and three-dimensional fermi surfaces and heavy cyclotron mass in the molecular conductor (DME-DCNQI)2Cu. Phys. Rev. B 1994, 50, 15597–15601. [Google Scholar] [CrossRef]
- Uji, S.; Shinagawa, H.; Terashima, T.; Yakabe, T.; Terai, Y.; Tokumoto, M.; Kobayashi, A.; Tanaka, H.; Kobayashi, H. Magnetic-field-induced superconductivity in a two-dimensional organic conductor. Nature 2001, 410, 908. [Google Scholar] [CrossRef] [PubMed]
- Sawa, H.; Tamura, M.; Aonuma, S.; Kinoshita, M.; Kato, R. Charge-transfer-controlled phase transition in a molecular conductor, (DME-DCNQI)2Cu –doping effect–. J. Phys. Soc. Jpn. 1994, 63, 4302–4305. [Google Scholar] [CrossRef]
- Fujiwara, H.; Kobayashi, H.; Fujiwara, E.; Kobayashi, A. An indication of magnetic-field-induced superconductivity in a bifunctional layered organic conductor, κ-(bets)2FeBr4. J. Am. Chem. Soc. 2002, 124, 6816–6817. [Google Scholar] [CrossRef]
- Coronado, E.; Galan-Mascaros, J.R.; Gomez-Garcia, C.J.; Laukhin, V. Coexistence of ferromagnetism and metallic conductivity in a molecule-based layered compound. Nature 2000, 408, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Sato, A.; Seki, M.; Saito, K.; Hiraki, K.-I.; Takahashi, T.; Kanoda, K.; Sorai, M. Spin-peierls transition of the quasi-one-dimensional electronic system (DMe-DCNQI) _ {2} M (M = Li, Ag) probed by heat capacity. Phys. Rev. B 2003, 68, 085112. [Google Scholar] [CrossRef]
- Coomber, A.T.; Beljonne, D.; Friend, R.H.; Brédas, J.L.; Charlton, A.; Robertson, N.; Underbill, A.E.; Kurmoo, M.; Day, P. Intermolecular interactions in the molecular ferromagnetic NH4Ni(mnt)2· H2O. Nature 1996, 380, 144. [Google Scholar] [CrossRef]
- Kato, R. Conducting metal dithiolene complexes: Structural and electronic properties. Chem. Rev. 2004, 104, 5319–5346. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Fujiwara, E.; Kobayashi, H. Single-component molecular metals with extended-ttf dithiolate ligands. Chem. Rev. 2004, 104, 5243–5264. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fujiwara, E.; Fujiwara, H.; Tanaka, H.; Otsuka, T.; Kobayashi, A.; Tokumoto, M.; Cassoux, P. Antiferromagnetic organic superconductors, bets2FeX4 (X=br, cl). Mol. Cryst. Liquid Cryst. 2002, 380, 139–144. [Google Scholar] [CrossRef]
- Pop, F.; Avarvari, N. Chiral metal-dithiolene complexes. Coord. Chem. Rev. 2017, 346, 20–31. [Google Scholar] [CrossRef]
- Dong, R.; Pfeffermann, M.; Liang, H.; Zheng, Z.; Zhu, X.; Zhang, J.; Feng, X. Large-area, free-standing, two-dimensional supramolecular polymer single-layer sheets for highly efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 2015, 54, 12058–12063. [Google Scholar] [CrossRef] [PubMed]
- Zarkadoulas, A.; Koutsouri, E.; Mitsopoulou, C.A. A perspective on solar energy conversion and water photosplitting by dithiolene complexes. Coord. Chem. Rev. 2012, 256, 2424–2434. [Google Scholar] [CrossRef]
- Kusamoto, T.; Nishihara, H. Zero-, one- and two-dimensional bis(dithiolato)metal complexes with unique physical and chemical properties. Coord. Chem. Rev. 2019, 380, 419–439. [Google Scholar] [CrossRef]
- Kato, R. Development of π-electron systems based on Mm(dmit)2] (M = Ni and Pd; dmit: 1,3-dithiole-2-thione-4,5-dithiolate) anion radicals. Bull. Chem. Soc. Jpn. 2014, 87, 355–374. [Google Scholar] [CrossRef]
- Naito, T.; Karasudani, T.; Mori, S.; Ohara, K.; Konishi, K.; Takano, T.; Takahashi, Y.; Inabe, T.; Nishihara, S.; Inoue, K. Molecular photoconductor with simultaneously photocontrollable localized spins. J. Am. Chem. Soc. 2012, 134, 18656–18666. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Karasudani, T.; Ohara, K.; Takano, T.; Takahashi, Y.; Inabe, T.; Furukawa, K.; Nakamura, T. Simultaneous control of carriers and localized spins with light in organic materials. Adv. Mater. 2012, 24, 6153–6157. [Google Scholar] [CrossRef]
- Noma, H.; Ohara, K.; Naito, T. [Cu(dmit)2]2− building block for molecular conductors and magnets with photocontrollable spin distribution. Chem. Lett. 2014, 43, 1230–1232. [Google Scholar] [CrossRef]
- Noma, H.; Ohara, K.; Naito, T. Direct control of spin distribution and anisotropy in cu-dithiolene complex anions by light. Inorganics 2016, 4, 7. [Google Scholar] [CrossRef]
- Naito, T. Development of a control method for conduction and magnetism in molecular crystals. Bull. Chem. Soc. Jpn. 2017, 90, 89–136. [Google Scholar] [CrossRef]
- Ray, K.; Weyhermüller, T.; Neese, F.; Wieghardt, K. Electronic structure of square planar bis(benzene-1,2-dithiolato)metal complexes [M(L)2]z (z = 2−, 1−, 0; M = Ni, Pd, Pt, Cu, Au): An experimental, density functional, and correlated ab initio study. Inorg. Chem. 2005, 44, 5345–5360. [Google Scholar] [CrossRef]
- Gewirth, A.A.; Cohen, S.L.; Schugar, H.J.; Solomon, E.I. Spectroscopic and theoretical studies of the unusual epr parameters of distorted tetrahedral cupric sites: Correlations to X-ray spectral features of core levels. Inorg. Chem. 1987, 26, 1133–1146. [Google Scholar] [CrossRef]
- Hoffmann, S.K.; Goslar, J.; Lijewski, S.; Zalewska, A. Epr and ese of cus4 complex in Cu(dmit)2: g-factor and hyperfine splitting correlation in tetrahedral cu–sulfur complexes. J. Magn. Reson. 2013, 236, 7–14. [Google Scholar] [CrossRef]
- Ozarowski, A.; Calzado, C.J.; Sharma, R.P.; Kumar, S.; Jezierska, J.; Angeli, C.; Spizzo, F.; Ferretti, V. Metal–metal interactions in trinuclear copper(II) complexes [Cu3(RCOO)4(H2tea)2] and binuclear [Cu2(RCOO)2(H2tea)2]. Syntheses and combined structural, magnetic, high-field electron paramagnetic resonance, and theoretical studies. Inorg. Chem. 2015, 54, 11916–11934. [Google Scholar] [CrossRef]
- Ozarowski, A. The zero-field-splitting parameter D in binuclear copper(II) carboxylates is negative. Inorg. Chem. 2008, 47, 9760–9762. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Jezierska, J.; Pombeiro, A.J.L.; Ozarowski, A. Copper(II) complexes with bulky n-substituted diethanolamines: High-field electron paramagnetic resonance, magnetic, and catalytic studies in oxidative cyclohexane amidation. Inorg. Chem. 2018, 57, 12384–12397. [Google Scholar] [CrossRef]
- Reger, D.L.; Pascui, A.E.; Foley, E.A.; Smith, M.D.; Jezierska, J.; Wojciechowska, A.; Stoian, S.A.; Ozarowski, A. Dinuclear metallacycles with single M–X–M bridges (X = Cl–, Br–; M = Fe(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II)): Strong antiferromagnetic superexchange interactions. Inorg. Chem. 2017, 56, 2884–2901. [Google Scholar] [CrossRef]
- Eisenberg, R.; Gray, H.B. Noninnocence in metal complexes: A dithiolene dawn. Inorg. Chem. 2011, 50, 9741–9751. [Google Scholar] [CrossRef]
- McCleverty, J.A. Metal 1,2-dithiolene and related complexes. In Progress in Inorganic Chemistry; Cotton, F.A., Ed.; Wiley Online Library: Hoboken, NJ, USA, 2007. [Google Scholar]
- Olk, R.-M.; Olk, B.; Dietzsch, W.; Kirmse, R.; Hoyer, E. The chemistry of 1,3-dithiole-2-thione-4,5-dithiolate (dmit). Coord. Chem. Rev. 1992, 117, 99–131. [Google Scholar] [CrossRef]
- Szilagyi, R.K.; Lim, B.S.; Glaser, T.; Holm, R.H.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Description of the ground state wave functions of ni dithiolenes using sulfur k-edge X-ray absorption spectroscopy. J. Am. Chem. Soc. 2003, 125, 9158–9169. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Rivera, J.; Maynau, D.; Calzado, C.J. Evaluation of the magnetic interactions in salts containing [ni(dmit)2]− radical anions. Chem. Mater. 2017, 29, 4317–4329. [Google Scholar] [CrossRef]
- Sarangi, R.; DeBeer George, S.; Rudd, D.J.; Szilagyi, R.K.; Ribas, X.; Rovira, C.; Almeida, M.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Sulfur k-edge X-ray absorption spectroscopy as a probe of ligand–metal bond covalency: Metal vs ligand oxidation in copper and nickel dithiolene complexes. J. Am. Chem. Soc. 2007, 129, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Stach, J.; Kirmse, R.; Dietzsch, W.; Olk, R.M.; Hoyer, E. Single-crystal EPR spectra of tetra-n-butylammonium bis(isotrithione-3,4-dithiolato)cuprate(II). Inorg. Chem. 1984, 23, 4779–4780. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciardi, G.; Baerends, E.J. Structural properties of M(dmit)2-based (M = Ni, Pd, Pt; dmit2− = 2-thioxo-1,3-dithiole-4,5-dithiolato) molecular metals. Insights from density functional calculations. Inorg. Chem. 1998, 37, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.K.; Goslar, J.; Lijewski, S.; Tadyszak, K.; Zalewska, A.; Jankowska, A.; Florczak, P.; Kowalak, S. EPR and UV-Vis study on solutions of Cu(II) dmit complexes and the complexes entrapped in zeolite Z and ZIF-Cu(IM)2. Microporous Mesoporous Mater. 2014, 186, 57–64. [Google Scholar] [CrossRef]
- Kirmse, R.; Stach, J.; Dietzsch, W.; Steimecke, G.; Hoyer, E. Single-crystal epr studies on nickel(iii), palladium(III), and platinum(III) dithiolene chelates containing the ligands isotrithionedithiolate, o-xylenedithiolate, and maleonitriledithiolate. Inorg. Chem. 1980, 19, 2679–2685. [Google Scholar] [CrossRef]
- Cabrero, J.; Calzado, C.J.; Maynau, D.; Caballol, R.; Malrieu, J.P. Metal-ligand delocalization in magnetic orbitals of binuclear complexes. J. Phys. Chem. A 2002, 106, 8146–8155. [Google Scholar] [CrossRef]
- Calzado, C.J.; Malrieu, J.P. Proposal of an extended t-j hamiltonian for high-t-c cuprates from ab initio calculations on embedded clusters. Phys. Rev. B 2001, 63. [Google Scholar] [CrossRef]
- Gellé, A.; Munzarová, M.L.; Lepetit, M.-B.; Illas, F. Role of dynamical polarization of the ligand-to-metal charge transfer excitations in ab initio determination of effective exchange parameters. Phys. Rev. B 2003, 68, 125103. [Google Scholar] [CrossRef]
- Tenti, L.; Maynau, D.; Angeli, C.; Calzado, C.J. Highly efficient perturbative plus variational strategy based on orthogonal valence bond theory for the evaluation of magnetic coupling constants. Application to the trinuclear Cu(ii) site of multicopper oxidases. Phys. Chem. Chem. Phys. 2016, 18, 18365–18380. [Google Scholar] [CrossRef] [PubMed]
- Giner, E.; Angeli, C. Spin density and orbital optimization in open shell systems: A rational and computationally efficient proposal. J. Chem. Phys. 2016, 144, 104104. [Google Scholar] [CrossRef] [PubMed]
- Giner, E.; Angeli, C. Metal-ligand delocalization and spin density in the CuCl2 and [CuCl4]2− molecules: Some insights from wave function theory. J. Chem. Phys. 2015, 143, 124305. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.A., Jr.; Rushbrooke, G.S.; Gilbert, H.E. High-temperature series expansions for the spin-1/2 heisenberg model by the method of irreducible representations of the symmetric group. Phys. Rev. 1964, 135, A1272–A1277. [Google Scholar] [CrossRef]
- Roos, B.O. The Complete Active Space Self-Consistent Field Method and Its Applications in Electronic Structure Calculations; John Wiley & Sons: Hoboken, NJ, USA, 1987. [Google Scholar]
- Andersson, K.; Per-Ake, M.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.-Å.; Roos, B.O.;; Serrano-Andrés, L. The multi-state caspt2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Malmqvist, P.-A.K.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (ras) state interaction approach with spin--orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Miralles, J.; Castell, O.; Caballol, R.; Malrieu, J.-P. Specific ci calculation of energy differences: Transition energies and bond energies. Chem. Phys. 1993, 172, 33–43. [Google Scholar] [CrossRef]
- Miralles, J.; Daudey, J.-P.; Caballol, R. Variational calculation of small energy differences. The singlet-triplet gap in [cu2cl6]2−. Chem. Phys. Lett. 1992, 198, 555–562. [Google Scholar] [CrossRef]
- Malrieu, J.P.; Caballol, R.; Calzado, C.J.; de Graaf, C.; Guihery, N. Magnetic interactions in molecules and highly correlated materials: Physical content, analytical derivation, and rigorous extraction of magnetic hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. New relativistic ano basis sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. Main group atoms and dimers studied with a new relativistic ano basis set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef]
- Maynau, D. Casdi Package Developed at the Laboratoire de Physique Quantique; Université Paul Sabatier: Toulouse, France, 1998. [Google Scholar]
- Ben Amor, N.; Maynau, D. Size-consistent self-consistent configuration interaction from a complete active space. Chem. Phys. Lett. 1998, 286, 211–220. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔE | λ | f | Dominant Component of the Wave Function * | Weight (%) | δCu | |
|---|---|---|---|---|---|---|
| X 2Ag | 0 | |2a00 22200| | 84 | 0.8909 | ||
| 1 2Au | 1.97 | 628.2 | 0.83 × 10−4 | ------------------ | −0.2843 | |
| 2 2Au | 2.25 | 551.2 | 0.738 | 54.4 | 0.8830 | |
| 3 2Au | 3.30 | 375.7 | 0.026 | 59.8 | 0.0501 | |
| 4 2Au | 3.46 | 358.7 | 0.014 | 58.6 | 0.4909 | |
| 5 2Au | 3.52 | 352.3 | 0.24 × 10−4 | ------------------ | −0.2852 | |
| 6 2Au | 4.05 | 306.5 | 0.48 × 10−2 | 87.9 | −0.0730 | |
| 7 2Au | 4.07 | 304.7 | 0.023 | 85.2 | 0.4231 | |
| 8 2Au | 4.50 | 275.7 | 0.15 × 10−5 | ------------------ | −0.2821 | |
| 9 2Au | 4.66 | 266.1 | 0.47 × 10−2 | 58.2 | 0.6138 | |
| 10 2Au | 4.73 | 262.08 | 0.015 | 55.6 | 0.0566 | |
| 11 2Au | 4.80 | 258.6 | 0.564 | 18.0 ** | 0.8357 | |
| 12 2Au | 5.36 | 231.4 | 0.28 × 10−2 | 19.1 ** 16.9 ** | 0.8023 | |
| 13 2Au | 5.44 | 227.7 | 0.72 × 10−4 | ------------------ | −0.1959 | |
| 14 2Au | 5.55 | 223.6 | 0.12 × 10−3 | 23.1 23.3 | 0.0431 | |
| 15 2Au | 5.89 | 210.6 | 0.64 × 10−5 | ------------------ | −0.2852 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapata-Rivera, J.; Calzado, C.J. Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study. Molecules 2019, 24, 1088. https://doi.org/10.3390/molecules24061088
Zapata-Rivera J, Calzado CJ. Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study. Molecules. 2019; 24(6):1088. https://doi.org/10.3390/molecules24061088
Chicago/Turabian StyleZapata-Rivera, Jhon, and Carmen J. Calzado. 2019. "Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study" Molecules 24, no. 6: 1088. https://doi.org/10.3390/molecules24061088
APA StyleZapata-Rivera, J., & Calzado, C. J. (2019). Light-Induced Control of the Spin Distribution on Cu–Dithiolene Complexes: A Correlated Ab Initio Study. Molecules, 24(6), 1088. https://doi.org/10.3390/molecules24061088