
Traceless Solid-Phase Synthesis of Ketones via Acid-Labile Enol Ethers: Application in the Synthesis of Natural Products and Derivatives


Abstract
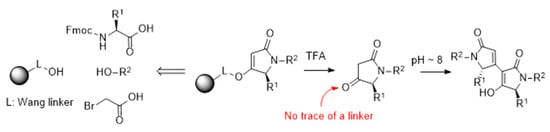
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Self-Condensation
2.3. Structure Determination
3. Conclusion
4. Experimental Procedures
4.1. General Information
4.2. Esterification with Fmoc–AA (Resins 2 and 4)
4.3. Reaction with Ns-Cl (Resin 3)
4.4. Fukuyama N-Alkylation (Resin 4)
4.5. Acylation with Bromoacetic Acid (Resin 5)
4.6. Preparation of the Triphenylphosphonium Salt (Resin 6)
4.7. Wittig Olefination (Resins 7)
4.8. Cleavage from the Resin (Compounds 8)
4.9. Self-Condensation (Compounds 9)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balkenhohl, F.; von dem Bussche-Hünnefeld, C.; Lansky, A.; Zechel, C. Combinatorial Synthesis of Small Organic Molecules. Angew. Chem. Int. Ed. 1996, 35, 2288–2337. [Google Scholar]
- Orzaez, M.; Mora, P.; Mondragon, L.; Perez-Paya, E.; Vicent, M.J. Solid-Phase Chemistry: A Useful Tool to Discover Modulators of Protein Interactions. Int. J. Peptide Res. Therap. 2007, 13, 281–293. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Meldal, M. Solid-Phase Synthesis of Complex and Pharmacologically Interesting Heterocycles. Curr. Opin. Drug Discovery Dev. 2009, 12, 798–810. [Google Scholar]
- Krchnak, V. Solid-Phase Synthesis of Nitrogenous Heterocycles; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Wang, S.-S. P-Alkoxybenzyl Alcohol Resin and P-Alkoxybenzyloxycarbonylhydrazide Resin for Solid Phase Synthesis of Protected Peptide Fragments. J. Am. Chem. Soc. 1973, 95, 1328–1333. [Google Scholar] [PubMed]
- James, I.W. Linkers for Solid Phase Organic Synthesis. Tetrahedron 1999, 55, 4855–4946. [Google Scholar] [CrossRef]
- Soural, M.; Hlavac, J.; Krchnak, V. Linkers for Solid-Phase Peptide Synthesis. In Solid-Phase Peptide Synthesis; Hughes, A.B., Ed.; Willey-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Krchnak, V.; Holladay, M.W. Solid Phase Heterocyclic Chemistry. Chem. Rev. 2002, 102, 61–91. [Google Scholar] [CrossRef]
- Winternheimer, D.J.; Shade, R.E.; Merlic, C.A. Methods for Vinyl Ether Synthesis. Synthesis 2010, 2010, 2497–2511. [Google Scholar]
- Murphy, P.J.; Lee, S.E. Recent Synthetic Applications of the Non-Classical Wittig Reaction. J. Chem. Soc. Perkin Trans. 1 1999, 21, 3049–3066. [Google Scholar]
- Sabitha, G.; Reddy, M.M.; Srinivas, D.; Yadov, J.S. Microwave Irradiation: Wittig Olefination of Lactones and Amides. Tetrahedron Lett. 1999, 40, 165–166. [Google Scholar] [CrossRef]
- Tsunoda, T.; Takagi, H.; Takaba, D.; Kaku, H.; Ito, S. Cyanomethylenetrimethylphosphorane, a Powerful Reagent for the Wittig Olefination of Esters, Lactones and Imides. Tetrahedron Lett. 2000, 41, 235–237. [Google Scholar] [CrossRef]
- Mortimore, M.; Kocienski, P. A New Synthesis of Spiroacetals Via Alkylidenation of Ester Carbonyls With Metal Carbene Complexes. Tetrahedron Lett. 1988, 29, 3357–3360. [Google Scholar]
- Macleod, C.; Hartley, R.C.; Hamprecht, D.W. Novel Functionalized Titanium(IV) Benzylidenes for the Traceless Solid- Phase Synthesis of Indoles. Org. Lett. 2002, 4, 75–78. [Google Scholar]
- Macleod, C.; McKiernan, G.J.; Guthrie, E.J.; Farrugia, L.J.; Hamprecht, D.W.; Macritchie, J.; Hartley, R.C. Synthesis of 2-Substituted Benzofurans and Indoles Using Functionalized Titanium Benzylidene Reagents on Solid Phase. J. Org. Chem. 2003, 68, 387–401. [Google Scholar] [PubMed]
- Hercouet, A.; Le Corre, M. Une Voie D’Acces Simple Aux Dihydro-2,3 Furannes Et Aux Dihydro-2,3 Pyrannes. Tetrahedron Lett. 1979, 20, 5–6. [Google Scholar] [CrossRef]
- Zhu, J.; Kayser, M.M. Synthesis of Enol Lactones Under a Solid/Liquid Phase Transfer Witting Reaction. Synth. Commun. 1994, 24, 1179–1186. [Google Scholar]
- Sakhautdinov, I.M.; Khalikov, I.G.; Galin, F.Z.; Egorov, V.A.; Lakeev, S.N.; Maidanova, I.O. The Comparative Study of Intramolecular Cyclization of Phthalimide Containing Sulfur and Phosphonium γ-Ylides. Bashk. Khim. Zh. 2007, 14, 96–99. [Google Scholar]
- Zhao, G.; Zhang, Q.; Zhou, H. Propargyl-Allenyl Isomerizations and Electrocyclizations for the Functionalization of Phosphonium Salts: One-Pot Synthesis of Polysubstituted Vinylbenzenes and Naphthalenes. Adv. Synth. Catal. 2013, 355, 3492–3496. [Google Scholar]
- Rahim, M.A.; Sasaki, H.; Saito, J.; Fujiwara, T.; Takeda, T. Intramolecular Carbonyl Olefination of Esters. Regioselective Preparation of Enol Ethers of Cyclic Ketones by the Titanocene(II)-Promoted Reaction of Alkyl ω,ω-Bis(Phenylthio)Alkanoates. Chem. Commun. (Cambridge, U.K.) 2001, 7, 625–626. [Google Scholar]
- Slade, R.M.; Phillips, M.A.; Berger, J.G. Application of an Almost Traceless Linker in the Synthesis of 2-Alkylthiobenzimidazole Combinatorial Libraries. Mol. Diversity 1998, 4, 215–219. [Google Scholar] [CrossRef]
- Hughes, I. Application of Polymer-Bound Phosphonium Salts As Traceless Supports for Solid Phase Synthesis. Tetrahedron Lett. 1996, 37, 7595–7598. [Google Scholar] [CrossRef]
- Desai, V.G.; Shet, J.B.; Tilve, S.G.; Mali, R.S. Intramolecular Wittig Reactions. A New Synthesis of Coumarins and 2-Quinolones. J. Chem. Res. Synop. 2003, 10, 628–629. [Google Scholar] [CrossRef]
- Schütznerova, E.; Pribylka, A.; Krchnak, V. Na-Amino Acid Containing Privileged Structures: Design, Synthesis and Use in Solid-Phase Peptide Synthesis. Org. Biomol. Chem. 2018, 16, 5359–5362. [Google Scholar] [CrossRef] [PubMed]
- Schütznerova, E.; Oliver, A.G.; Pribylka, A.; Krchnak, V. Solid-Phase Synthesis of Tetramic Acid Via Resin-Bound Enol Ethers As a Privileged Scaffold in Drug Discovery. Adv. Synth. Catal. 2018, 360, 3693–3699. [Google Scholar]
- Hill, C.M.; Prigmore, R.M.; Moore, G.J. Grignard Reagents and Unsaturated Ethers. IV. The Synthesis and Reaction of Several Vinyl Ethers With Grignard Reagents. J. Am. Chem. Soc. 1955, 77, 352–354. [Google Scholar]
- Fife, T.H. Vinyl Ether Hydrolysis. The Facile General Acid Catalyzed Conversion of 2-Ethoxy-1-Cyclopentene-1-Carboxylic Acid to Cyclopentanone. J. Am. Chem. Soc. 1965, 87, 1084–1089. [Google Scholar] [PubMed]
- Fedor, L.R.; McLaughlin, J. Vinyl Ether Hydrolysis. Specific Acid Catalyzed Hydrolysis of 4-Methoxy-3-Buten-2-One. J. Am. Chem. Soc. 1969, 91, 3594–3597. [Google Scholar] [CrossRef] [PubMed]
- Bergman, N.A.; Halvarsson, T. Hydrolysis of the Vinyl Ether Functional Group in a Model for Prostacyclin in Which the Carboxyl Group Has Been Replaced by a Pyridine Ring. J. Org. Chem. 1989, 54, 2137–2142. [Google Scholar] [CrossRef]
- Cheon, C.H.; Yamamoto, H. A Brönsted Acid Catalyst for the Enantioselective Protonation Reaction. J. Am. Chem. Soc. 2008, 130, 9246–9247. [Google Scholar] [PubMed]
- Cheon, C.H.; Kanno, O.; Toste, F.D. Chiral Brönsted Acid From a Cationic Gold(I) Complex: Catalytic Enantioselective Protonation of Silyl Enol Ethers of Ketones. J. Am. Chem. Soc. 2011, 133, 13248–13251. [Google Scholar] [CrossRef]
- Liu, M.; Hyder, Z.; Sun, Y.; Tang, W.; Xu, L.; Xiao, J. Efficient Synthesis of Alkyl Aryl Ketones & Ketals Via Palladium-Catalyzed Regioselective Arylation of Vinyl Ethers. Org. Biomol. Chem. 2010, 8, 2012–2015. [Google Scholar]
- Huang, W.; Rong, H.Y.; Xu, J. Cyclic a-Alkoxyphosphonium Salts From (2-(Diphenylphosphino)Phenyl)Methanol and Aldehydes and Their Application in Synthesis of Vinyl Ethers and Ketones Via Wittig Olefination. J. Org. Chem. 2015, 80, 6628–6638. [Google Scholar] [PubMed]
- Mo, X.; Li, Q.; Ju, J. Naturally Occurring Tetramic Acid Products: Isolation, Structure Elucidation and Biological Activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar]
- Bai, W.-J.; Lu, C.; Wang, X. Recent Advances in the Total Synthesis of Tetramic Acid-Containing Natural Products. J. Chem. 2016. [Google Scholar] [CrossRef]
- Royles, B.J.L. Naturally Occurring Tetramic Acids: Structure, Isolation, and Synthesis. Chem. Rev. 1995, 95, 1981–2001. [Google Scholar] [CrossRef]
- Athanasellis, G.; Igglessi-Markopoulou, O.; Markopoulos, J. Tetramic and Tetronic Acids As Scaffolds in Bioinorganic and Bioorganic Chemistry. Bioinorg. Chem. Appl. 2010. [Google Scholar] [CrossRef]
- Fukuyama, T.; Jow, C.-K.; Cheung, M. 2- and 4-Nitrobenzenesulfonamides: Exceptionally Versatile Means for Preparation of Secondary Amines and Protection of Amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar] [CrossRef]
- Heinicke, G.W.; Morella, A.M.; Orban, J.; Prager, R.H.; Ward, A.D. Central-Nervous-System Active Compounds .XVI. Some Chemistry of 6-Oxo Caprolactams Derived From an Enamine Ring-Expansion Synthesis. Aust. J. Chem. 1985, 38, 1847–1856. [Google Scholar] [CrossRef]
- Jeong, Y.-C.; Moloney, M.G. Tetramic Acids as Scaffolds: Synthesis, Tautomeric and Antibacterial Behaviour. Synlett 2009, 15, 2487–2491. [Google Scholar]
- Wilk, W.; Zimmermann, T.J.; Kaiser, M.; Waldmann, H. Principles, Implementation, and Application of Biology-Oriented Synthesis (BIOS). Biol. Chem. 2010, 391, 491–497. [Google Scholar]
- Kaiser, M.; Wetzel, S.; Kumar, K.; Waldmann, H. Biology-Inspired Synthesis of Compound Libraries. Cell. Mol. Life Sci. 2008, 65, 1186–1201. [Google Scholar] [CrossRef]
- Wetzel, S.; Klein, K.; Renner, S.; Rauh, D.; Oprea, T.I.; Mutzel, P.; Waldmann, H. Interactive Exploration of Chemical Space With Scaffold Hunter. Nat. Chem. Biol. 2009, 5, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-Oriented Synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Van Hattum, H.; Waldmann, H. Biology-Oriented Synthesis: Harnessing the Power of Evolution. J. Am. Chem. Soc. 2014, 136, 11853–11859. [Google Scholar] [CrossRef] [PubMed]
- Krchnak, V.; Padera, V. The Domino Blocks: A Simple Solution for Parallel Solid Phase Organic Synthesis. Bioorg. Med. Chem. Lett. 1998, 22, 3261–3264. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Route | R1 | R2 | Purity of 8 (%) | Yield of 8 (%) b | Yield of 9 (%) c |
|---|---|---|---|---|---|---|---|
| 1 | {1,1} | I | H | Me | 85 | NI d | 47 |
| 2 | {1,2} | II | H | PhthN(CH2)2 | 81 | 70 | 53 |
| 3 | {2,-} | I | Hyp(Bzl) a | 81 | NI d | 37 | |
| 4 | {3,-} | I | Idc a | 52 | 31 | NT e | |
| 5 | {4,1} | I | CH2C6H4OH | Me | 77 | 25 | NT e |
| 6 | {5,3} | II | Me | Bn | 56 | NI d | 23 |
| 7 | {6,2} | II | Bn | PhthN(CH2)2 | 72 | 30 | 49 |
| 8 | {6,4} | II | Bn | CH2CCH | 88 | 21 | NT e |
| 9 | {7,2} | II | CH2OH | PhthN(CH2)2 | 68 | 52 | NT e |
| 10 | {8,2} | II | (CH2)2CO2H | PhthN(CH2)2 | 52 | 21 | NT e |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schütznerová, E.; Krchňáková, A.; Krchňák, V. Traceless Solid-Phase Synthesis of Ketones via Acid-Labile Enol Ethers: Application in the Synthesis of Natural Products and Derivatives. Molecules 2019, 24, 1406. https://doi.org/10.3390/molecules24071406
Schütznerová E, Krchňáková A, Krchňák V. Traceless Solid-Phase Synthesis of Ketones via Acid-Labile Enol Ethers: Application in the Synthesis of Natural Products and Derivatives. Molecules. 2019; 24(7):1406. https://doi.org/10.3390/molecules24071406
Chicago/Turabian StyleSchütznerová, Eva, Anna Krchňáková, and Viktor Krchňák. 2019. "Traceless Solid-Phase Synthesis of Ketones via Acid-Labile Enol Ethers: Application in the Synthesis of Natural Products and Derivatives" Molecules 24, no. 7: 1406. https://doi.org/10.3390/molecules24071406
APA StyleSchütznerová, E., Krchňáková, A., & Krchňák, V. (2019). Traceless Solid-Phase Synthesis of Ketones via Acid-Labile Enol Ethers: Application in the Synthesis of Natural Products and Derivatives. Molecules, 24(7), 1406. https://doi.org/10.3390/molecules24071406