Simultaneous Determination of Bufalin and Its Nine Metabolites in Rat Plasma for Characterization of Metabolic Profiles and Pharmacokinetic Study by LC–MS/MS



Abstract
:
1. Introduction
2. Materials and Methods
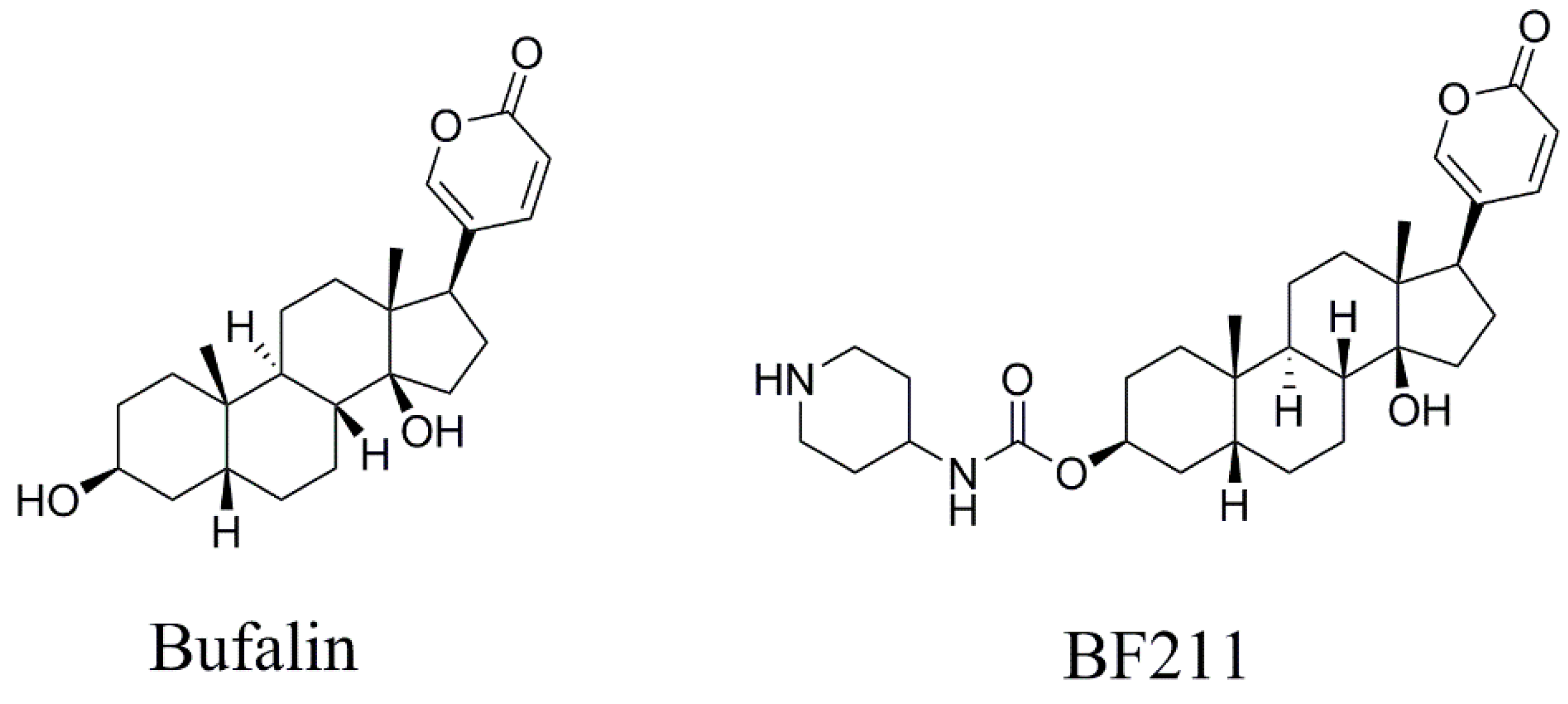
2.1. Chemicals and Reagents
2.2. UPLC–QTOF/MS Instruments and Conditions
2.3. LC–MS/MS Instruments and Conditions
2.4. Preparation of Calibration Standards and Quality Control (QC) Samples
2.5. Sample Preparation
2.6. LC–MS/MS Method Validation
2.6.1. Selectivity
2.6.2. Accuracy and Precision
2.6.3. Recovery and Matrix Effect
2.6.4. Stability
2.7. Characterization of Metabolic Profiles and Pharmacokinetic Study in Rats
3. Results and Discussion
3.1. Identification of Metabolites of Bufalin
3.2. LC–MS/MS Condition Optimization
3.3. Sample Preparation
3.4. Method Validation
3.5. Characterization of Metabolic Profiles and Pharmacokinetic Study in Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IS | internal standard |
| MRM | multiple reaction monitoring |
| LOQ | limit of quantitation |
| VB | Venenum Bufonis |
| TCM | Traditional Chinese Medicine |
| QC | quality control |
| LLOQ | lower limit of quantification |
| RSD | relative standard deviation. |
References
- China Pharmacopoeia Committee. 2015 Edition of Chinese Pharmacopoeia; Chemical Industry Press: Beijing, China, 2015; pp. 383–384. [Google Scholar]
- Wansapura, A.N.; Lasko, V.; Xie, Z.; Fedorova, O.V.; Bagrov, A.Y.; Lingrel, J.B.; Lorenz, J.N. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive alpha1 Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 1833–1839. [Google Scholar] [CrossRef]
- Gao, H.M.; Popescu, R.; Kopp, B.; Wang, Z.M. Bufadienolides and their antitumor activity. Nat. Prod. Rep. 2011, 28, 953–969. [Google Scholar] [CrossRef]
- Slingerland, M.; Cerella, C.; Guchelaar, H.J.; Diederich, M.; Gelderblom, H. Cardiac glycosides in cancer therapy: From preclinical investigations towards clinical trials. Invest New Drugs. 2013, 31, 1087–1094. [Google Scholar] [CrossRef]
- Ye, M.; Guo, H.; Guo, H.Z.; Han, J.; Guo, D.A. Simultaneous determination of cytotoxic bufadienolides in the Chinese medicine ChanSu by high-performance liquid chromatography coupled with photodiode array and mass spectrometry detections. J. Chromatogr. B. 2006, 838, 86–95. [Google Scholar] [CrossRef]
- Zhu, Z.T.; Sun, H.Z.; Ma, G.Y.; Wang, Z.H.; Li, E.Z.; Liu, Y.Y.; Liu, Y.P. Bufalin induces lung cancer cell apoptosis via the inhibition of PI3K/Akt pathway. Int. J. Mol. Sci. 2012, 13, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.L.; Han, J.W.; Zhai, B.; Ming, X.D.; Zhuang, L.W.; Liu, Y.; Pan, S.H.; Liu, T.F. Blocking autophagy enhances the apoptosis effect of bufalin on human hepatocellular carcinoma cells through endoplasmic reticulum stress and JNK activation. Apoptosis 2014, 19, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.P.; Yu, C.S.; Yu, C.C.; Yang, J.S.; Chiang, J.H.; Lu, C.C.; Huang, H.Y.; Tang, N.Y.; Yang, J.H.; Huang, A.C.; et al. Triggering apoptotic death of human malignant melanoma A375.S2 cells by bufalin: Involvement of caspase cascade-dependent and independent mitochondrial signaling pathways. Evid. Based Complement. Alternat. Med 2012, 591241. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, G.Z. Poisoning by toxic animals in China-18 autopsy case studies and a comprehensive literature review. Forensic Sci. Int. 2013, e12–e23. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.M.; Medeiros, M.S.; Riani Costa, C.C.M.; Camplesi, A.C.; Sakate, M. Toad poisoning in three dogs: Case reports. J. Venom. Anim. Toxins Incl. Trop Dis. 2009, 15, 789–798. [Google Scholar] [CrossRef]
- Ning, J. Studies on the In Vivo Metabolism of Cinobufagin in Rat and the Comparative Metabolic Species Difference In Vitro. Master’s Thesis, Dalian Medical university, Dalian, China, 2011. [Google Scholar]
- Ning, J. The In Vitro Metabolism Investigation of Bufadienolides, Active Constisuents of Chansu. Ph.D. Thesis, Dalian Medical university, Dalian, China, 2017. [Google Scholar]
- Han, L.; Wang, H.J.; Si, N.; Ren, W.; Gao, B.; Li, Y.; Yang, J.; Xu, M.; Zhao, H.Y.; Bian, B.L. Metabolites profiling of 10 bufadienolides in human liver microsomes and their cytotoxicity variation in HepG2 cell. Anal. Bioanal. Chem. 2016, 408, 2485–2495. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Hou, J.; Wang, P.; Wu, J.J.; Dai, Z.R.; Zou, L.W.; Li, W.; Ge, G.B.; Ma, X.C.; Yang, L. Interspecies variation in phase I metabolism of bufalin in hepatic microsomes from mouse, rat, dog, minipig, monkey, and human. Xenobiotica 2015, 45, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Miyashiro, Y.; Nishio, T. Characterization of in vitro metabolites of toad venom using high-performance liquid chromatography and liquid chromatography-mass spectrometry. Biomed. Chromatogr. 2006, 20, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.L.; Dong, P.P.; Sun, X.H.; Deng, S.; Zhang, N.; Wang, C.; Huo, X.K.; Li, Y.; Lan, R.; Chen, L.; et al. Identification of the hydroxylated derivatives of bufalin: Phase I metabolites in rats. J. Asian Nat. Prod. Res. 2016, 18, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.N.; Zhou, X.X.; Cao, W.; Bi, L.L.; Zhang, Y.F.; Yang, Q.; Wang, S.W. Improved antitumor efficacy and pharmacokinetics of bufalin via PEGylated liposomes. Nanoscale Res. Lett. 2017, 12, 585. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Xiao, Z.Y.; Chen, Y.J.; Lei, M.; Meng, Y.H.; Guo, D.A.; Liu, X.; Hu, L.H. Synthesis and structure-activity relationships study of cytotoxic bufalin 3-nitrogen-containing-ester derivatives. Steroids 2013, 78, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Huang, C.R.; Xue, L.; Xu, Q.Q.; Xia, W.; Li, X.; Miao, L.Y. Simultaneous determination of bentysrepinine (Y101) and its metabolites M8 and M9 in human plasma by UPLC–MS/MS and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2018, 50, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Q.; Siard, M.; Adams, A.; Keowen, M.L.; Miller, T.K.; Garza, F., Jr.; Andrews, F.M.; Seeram, N.P. Simultaneous quantification of free curcuminoids and their metabolites in equine plasma by LC-ESI–MS/MS. J. Pharm. Biomed. Anal. 2018, 154, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Peng, C.C.; Jiang, P.; Fu, P.; Tao, J.F.; Han, L.; Huang, H.M.; Chang, W.L.; Li, L.; Zhang, W.D.; et al. Simultaneous determination of seven bufadienolides in rat plasma after oral administration of Shexiang Baoxin Pill by liquid chromatography-electrospray ionization-tandem mass spectrometry: Application to a pharmacokinetic study. J. Chromatogr. B. 2014, 967, 255–263. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds bufalin and BF211 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Component Name | Pathway | Formula | M + H (m/z) | Retention Time (RT, min) | Fragments |
|---|---|---|---|---|---|---|
| 1 | 3-epi-bufalin | Isomerization | C24H34O4 | 387.2527 | 15.56 | 369.24; 351.23; 333.22; 255.21 |
| 2 | Unknown | Bufalin-O(cleavage) + H2 | C24H36O3 | 373.2735 | 18.63 | 355.26; 337.25; 255.21 |
| 3 | Unknown | Bufalin-O(cleavage) + H2 | C24H36O3 | 373.2737 | 14.6 | 355.26; 337.25; 255.21 |
| 4 | Unknown | Bufalin-O(cleavage) + H2 | C24H36O3 | 373.2739 | 13.95 | 355.26; 337.25; 255.21 |
| 5 | Unknown | Bufalin + 2 × H2 | C24H38O4 | 391.2830 | 15.24 | 373.27; 355.26; 337.25 |
| 6 | Unknown | Bufalin + 2 × H2 | C24H38O4 | 391.2833 | 20.84 | 373.27; 355.26; 337.25 |
| 7 | Hydroxylation-bufalin | Bufalin + O | C24H34O5 | 403.2473 | 9.65 | 385.24; 367.23; 349.22; 331.21; 253.19 |
| 8 | Hydroxylation-bufalin | Bufalin + O | C24H34O5 | 403.2470 | 7.58 | 385.24; 367.23; 349.22; 331.21; 253.19 |
| 9 | Hydroxylation-bufalin | Bufalin + O | C24H34O5 | 403.2471 | 6.75 | 385.24; 367.23; 349.22; 331.21; 253.19 |
| 10 | Hydroxylation-bufalin | Bufalin + O | C24H34O5 | 403.2451 | 10.57 | 385.24; 367.23; 349.22; 331.21; 253.19 |
| 11 | Unknown | Unknown | C24H34O5 | 403.2451 | 7.05 | No found |
| 12 | Unknown | Unknown | C24H34O5 | 403.2492 | 9.87 | No found |
| 13 | Dihydroxy-bufalin | Bufalin + 2 × O | C24H34O6 | 419.2424 | 7.01 | 401.24; 383.22; 365.21; 347.20 |
| 14 | Unknown | bufalin-O + 2 × (-H2) + C2H2O | C26H32O4 | 409.2345 | 15.55 | 369.24; 351.23 |
| Metabolites | ESI Mode | QTOF/MS | MS/MS | ||
|---|---|---|---|---|---|
| Precursor ion (M + H) | Fragments | Precursor Ion | Product Ion | ||
| 3-epi-bufalin | ES+ | 387.2527 | 369.24; 351.23; 333.22; 255.21 | 387.40 | 369.60 |
| Dihydroxylated bufalin | ES+ | 419.2424 | 401.24; 383.22; 365.21; 347.20 | 419.24 | 365.21 |
| Hydroxylated bufalin | ES+ | 403.2473 | 385.24; 367.23; 349.22; 331.21; 253.19 | 403.25 | 349.22 |
| Hydroxylated bufalin | ES+ | 403.2470 | 385.24; 367.23; 349.22; 331.21; 253.19 | 403.25 | 349.22 |
| Hydroxylated bufalin | ES+ | 403.2471 | 385.24; 367.23; 349.22; 331.21; 253.19 | 403.25 | 349.22 |
| Hydroxylated bufalin | ES+ | 403.2451 | 385.24; 367.23; 349.22; 331.21; 253.19 | 403.25 | 349.22 |
| Analyte | Q1 Mass (Da) | Q3 Mass (Da) | DP (Volts) | CE (Volts) |
|---|---|---|---|---|
| Bufalin | 387.4 | 369.6 | 115 | 41 |
| BF211 | 513.7 | 145.3 | 78 | 26 |
| Analyte. | Spiked Conc. (ng/mL) | Intra-Batch (n = 6) | Inter-Batch (n = 6 × 3) | ||||
|---|---|---|---|---|---|---|---|
| Measured Conc. (mean ± SD, ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Measured Conc. (mean ± SD, ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | ||
| Bufalin | 1.00 | 1.08 ± 0.13 | 12.04 | 8.28 | 1.06 ± 0.11 | 10.28 | 6.09 |
| 3.00 | 3.26 ± 0.17 | 5.10 | 8.61 | 3.26 ± 0.18 | 5.66 | 8.61 | |
| 15.00 | 16.42 ± 0.51 | 3.09 | 9.44 | 16.39 ± 0.69 | 4.19 | 9.30 | |
| 80.00 | 75.68 ± 3.66 | 4.83 | −5.40 | 82.23 ± 5.95 | 7.23 | 2.79 | |
| Analytes | Nominal Conc. (ng/mL) | Room Temperature Stability for 4 h | Autosampler Stability for 24 h | Freeze–Thaw Stability for Three Cycles at −80 °C | The Long-Term Stability at −80 °C for 25 Days | ||||
|---|---|---|---|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | ||
| Bufalin | 3.00 | 6.9 | 7.6 | 8.7 | −5.1 | 6.0 | 4.0 | 6.2 | 6.0 |
| 80.0 | 5.6 | 2.8 | 5.0 | −11.4 | −0.9 | −4.0 | 4.8 | −6.0 | |
| Parameters | Cmax (ng/mL) | Tmax (h) | t1/2 (h) | AUC0-t (ug/L∗h) | MRT0-t (h) |
|---|---|---|---|---|---|
| Bufalin | 14.722 ± 4.681 | 0.25 ± 0.00 | 5.70 ± 3.06 | 37.31 ± 9.54 | 3.23 ± 0.37 |
| 3-epi-bufalin | 963.06 ± 284.76 | 0.42 ± 0.14 | 7.74 ± 3.87 | 2214.18 ± 460.31 | 3.21 ± 0.24 |
| Hydroxylated bufalin (RT = 1.29 min) | 57.37 ± 13.41 | 0.58 ± 0.14 | 2.23 ± 0.36 | 144.78 ± 23.41 | 2.41 ± 0.07 |
| Hydroxylated bufalin (RT = 1.63 min) | 13.90 ± 4.64 | 0.58 ± 0.14 | 1.90 ± 0.37 | 34.45 ± 8.82 | 2.41 ± 0.20 |
| Hydroxylated bufalin (RT = 2.02 min) | 299.56 ± 112.89 | 0.50 ± 0.00 | 3.13 ± 1.50 | 643.98 ± 173.93 | 2.46 ± 0.17 |
| Hydroxylated bufalin (RT = 3.42 min) | 107.36 ± 44.57 | 0.50 ± 0.00 | 2.32 ± 0.71 | 238.03 ± 64.08 | 2.30 ± 0.15 |
| Hydroxylated bufalin (RT = 3.81 min) | 15.28 ± 3.86 | 2.67 ±1.16 | 12.39 ± 9.22 | 83.59 ± 13.61 | 4.06 ± 0.17 |
| Dihydroxylated bufalin (RT = 0.84 min) | 6.14 ± 1.72 | 0.83 ± 0.29 | 2.49 ± 1.05 | 18.80 ± 2.74 | 2.84 ± 0.16 |
| Dihydroxylated bufalin (RT = 1.59 min) | 14.68 ± 6.52 | 1.67 ± 0.58 | 5.81 ± 3.68 | 72.20 ± 22.72 | 3.66 ± 0.14 |
| Dihydroxylated bufalin (RT = 2.68 min) | 3.25 ± 1.68 | 1.17 ± 0.76 | 2.03 ± 0.67 | 11.74 ± 5.11 | 2.91 ± 0.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, W.; Yu, Y.; Wang, X.; Yang, L.; Zhang, H.; Ji, H.; Li, Z.; Hou, J.; Wu, W.; Guo, D. Simultaneous Determination of Bufalin and Its Nine Metabolites in Rat Plasma for Characterization of Metabolic Profiles and Pharmacokinetic Study by LC–MS/MS. Molecules 2019, 24, 1662. https://doi.org/10.3390/molecules24091662
Wei W, Yu Y, Wang X, Yang L, Zhang H, Ji H, Li Z, Hou J, Wu W, Guo D. Simultaneous Determination of Bufalin and Its Nine Metabolites in Rat Plasma for Characterization of Metabolic Profiles and Pharmacokinetic Study by LC–MS/MS. Molecules. 2019; 24(9):1662. https://doi.org/10.3390/molecules24091662
Chicago/Turabian StyleWei, Wenlong, Yang Yu, Xia Wang, Linhui Yang, Hang Zhang, Hongjian Ji, Zhenwei Li, Jinjun Hou, Wanying Wu, and Dean Guo. 2019. "Simultaneous Determination of Bufalin and Its Nine Metabolites in Rat Plasma for Characterization of Metabolic Profiles and Pharmacokinetic Study by LC–MS/MS" Molecules 24, no. 9: 1662. https://doi.org/10.3390/molecules24091662
APA StyleWei, W., Yu, Y., Wang, X., Yang, L., Zhang, H., Ji, H., Li, Z., Hou, J., Wu, W., & Guo, D. (2019). Simultaneous Determination of Bufalin and Its Nine Metabolites in Rat Plasma for Characterization of Metabolic Profiles and Pharmacokinetic Study by LC–MS/MS. Molecules, 24(9), 1662. https://doi.org/10.3390/molecules24091662