
Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles



Abstract
:1. Introduction
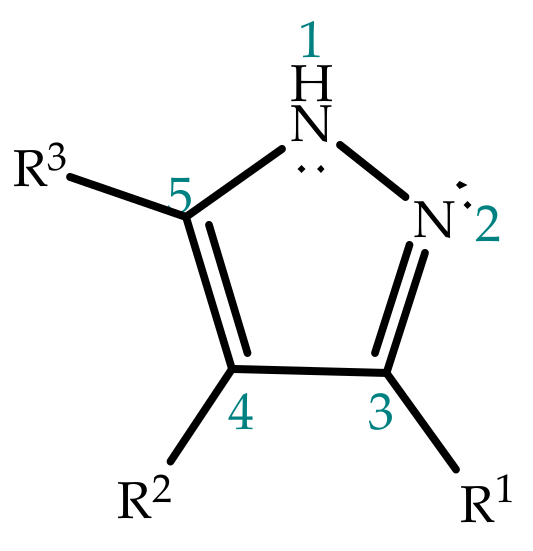
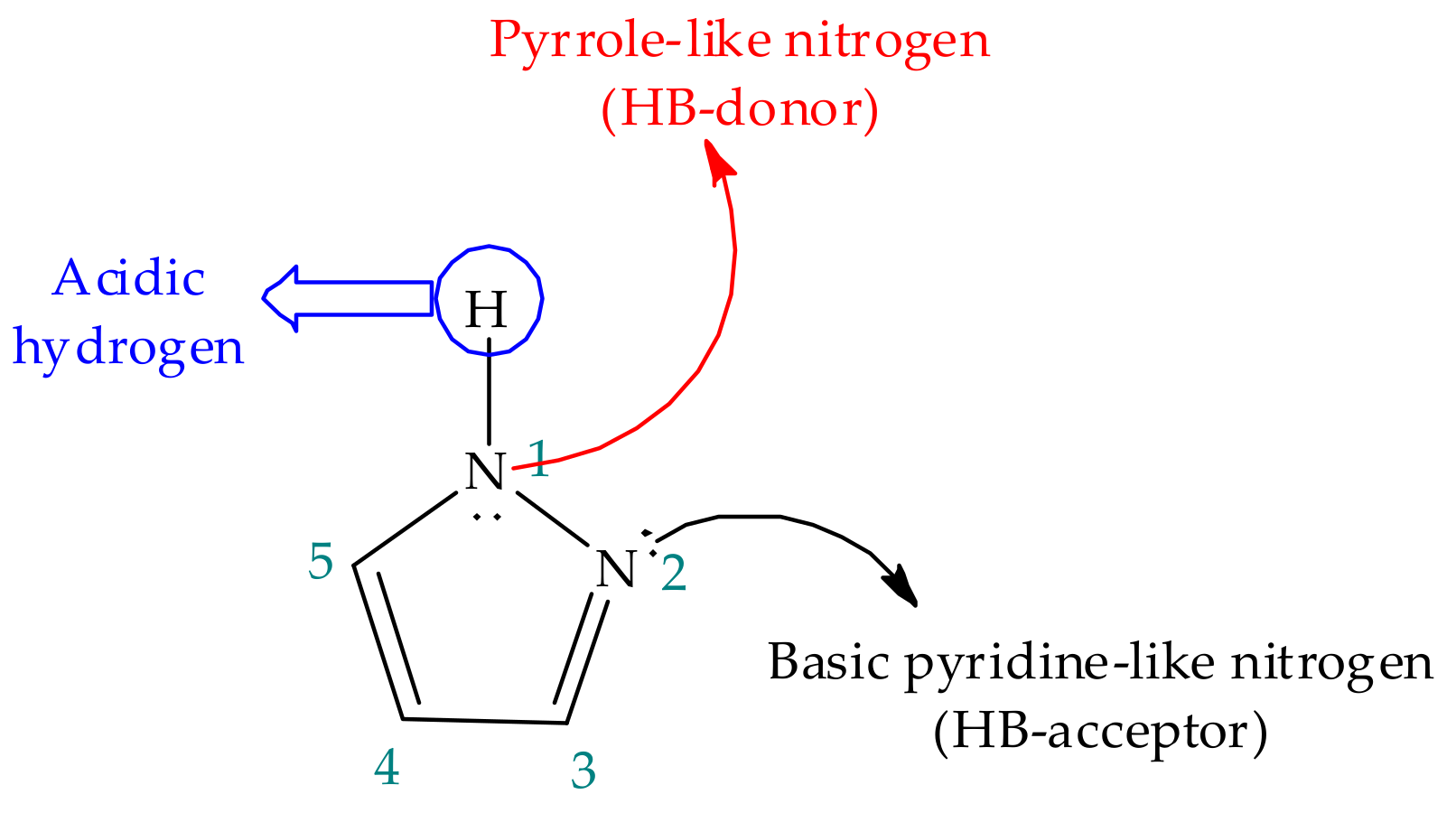
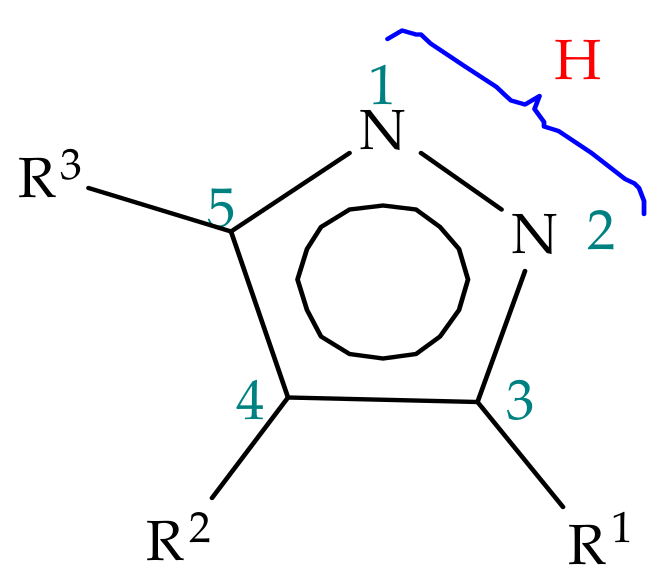
2. Pyrazole Properties
3. Tautomerism in Pyrazoles
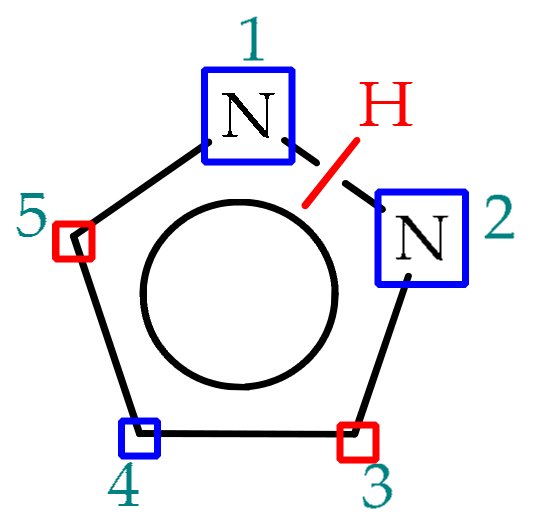
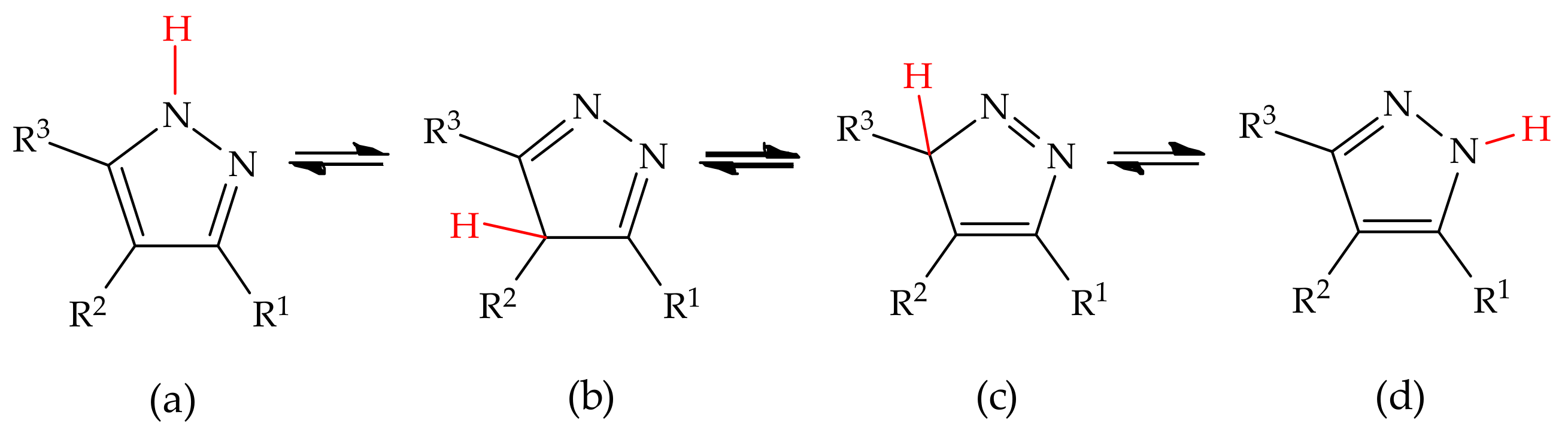
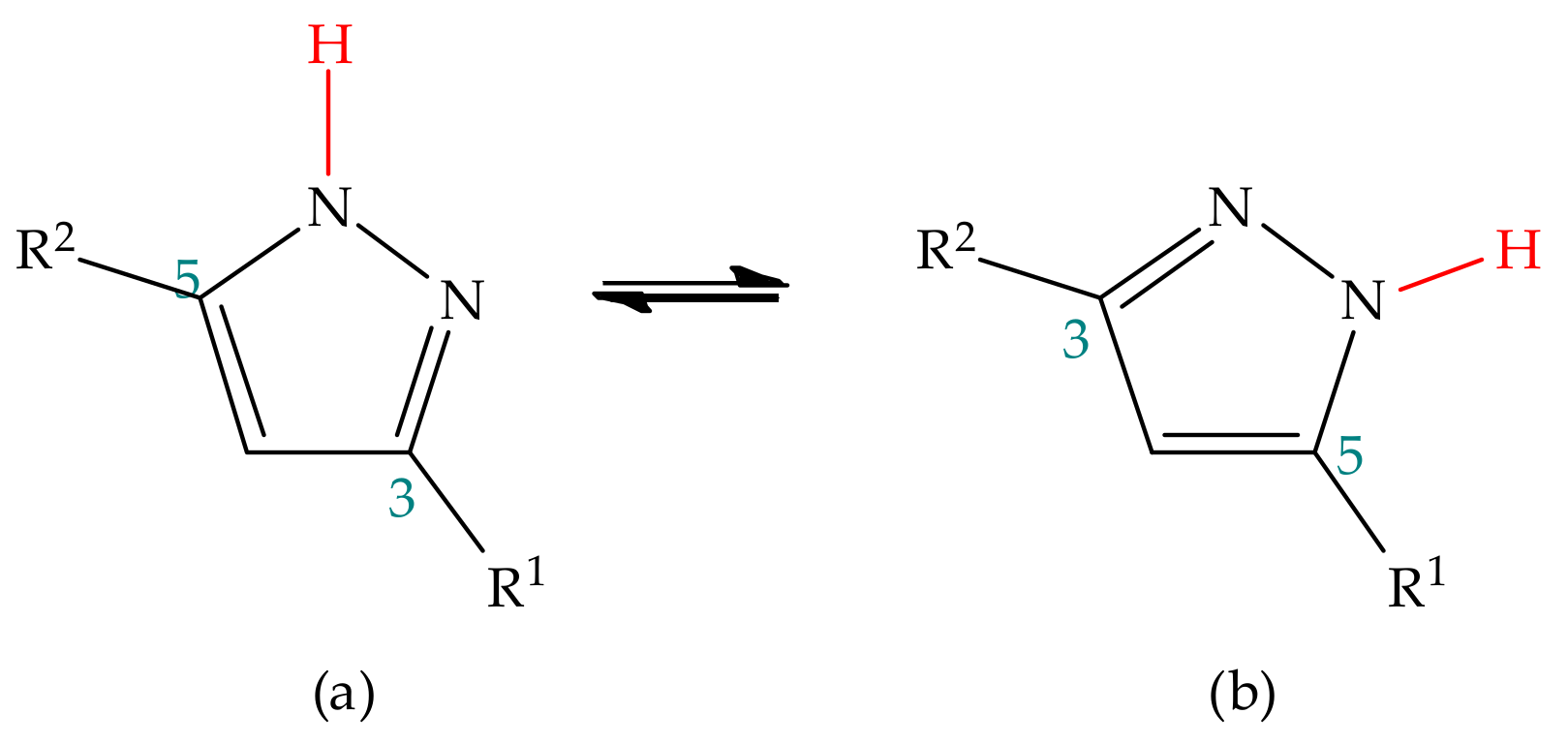
3.1. Annular Prototropic Tautomerism in Pyrazoles
3.1.1. Theoretical Studies
Tautomer Ratio
Substituent Effects
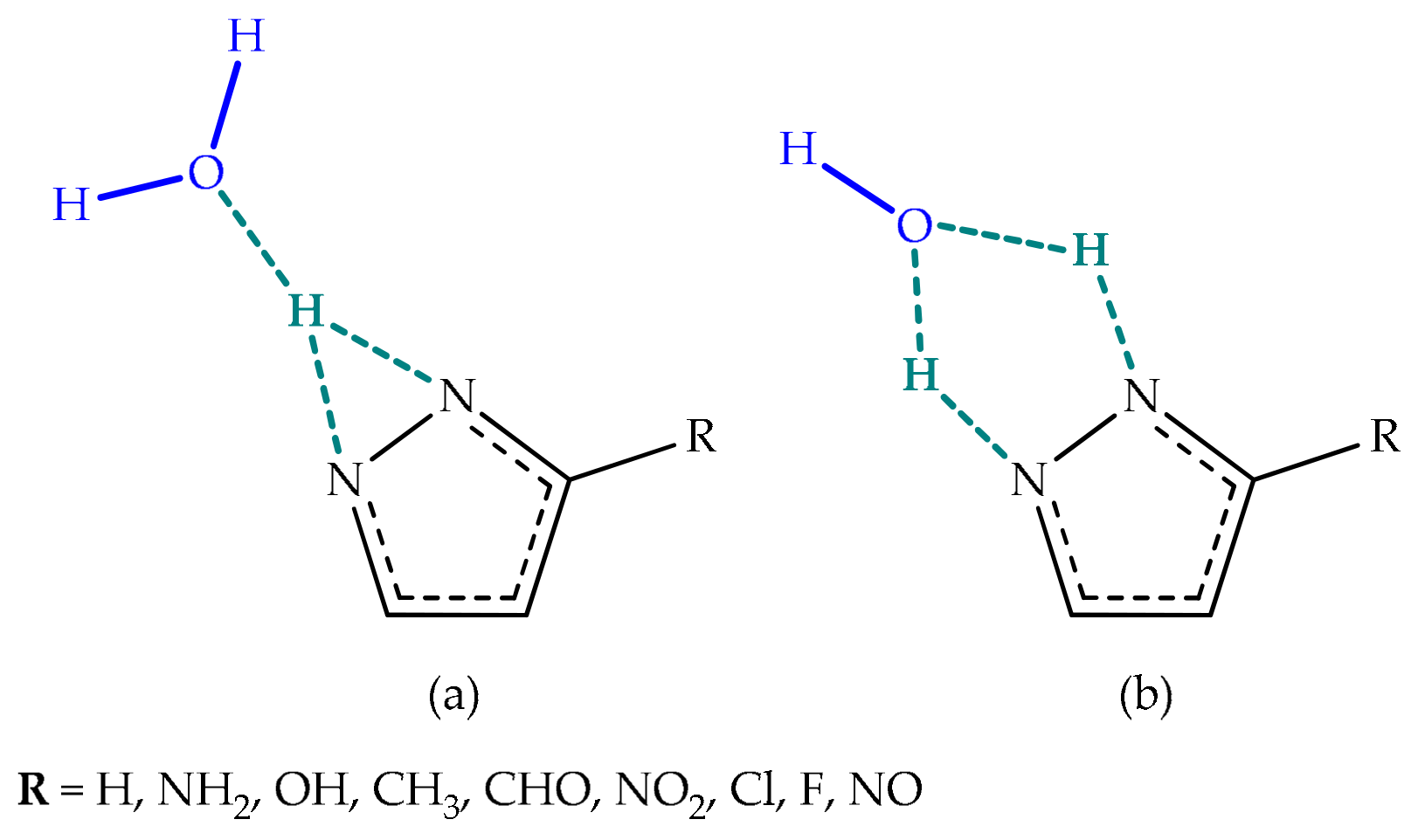
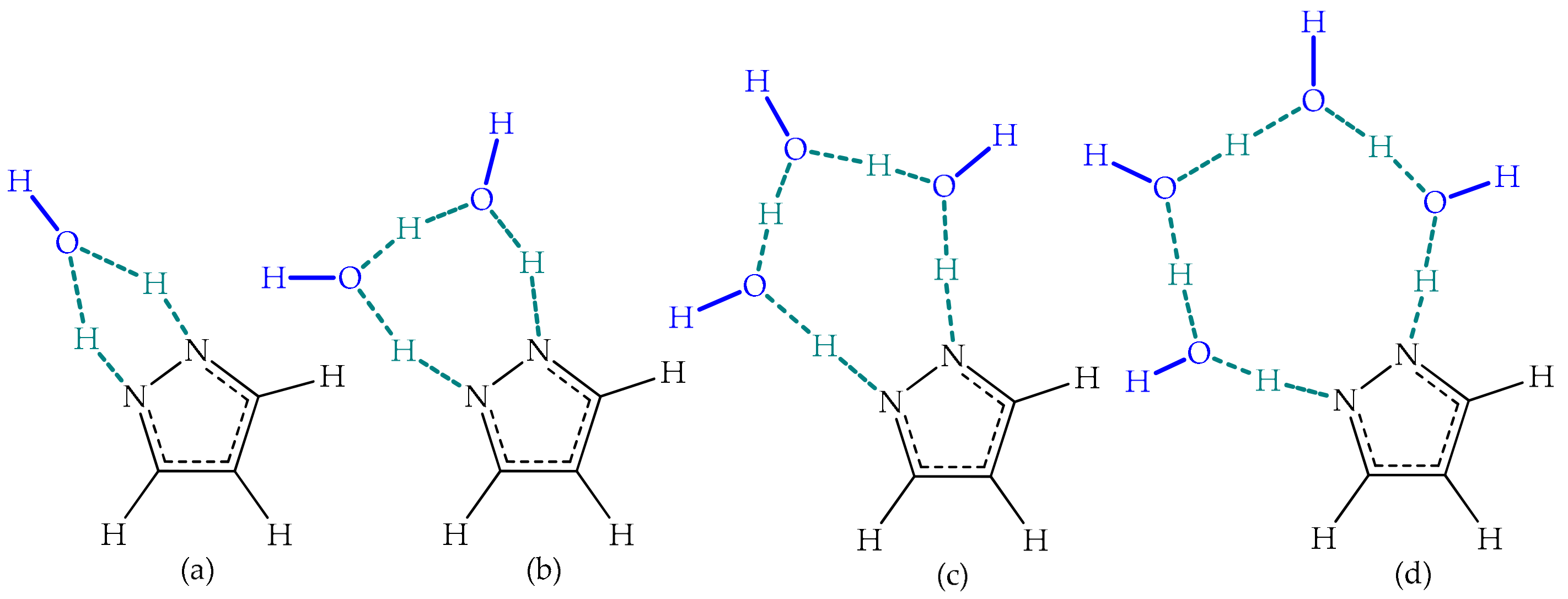
Environmental Effects
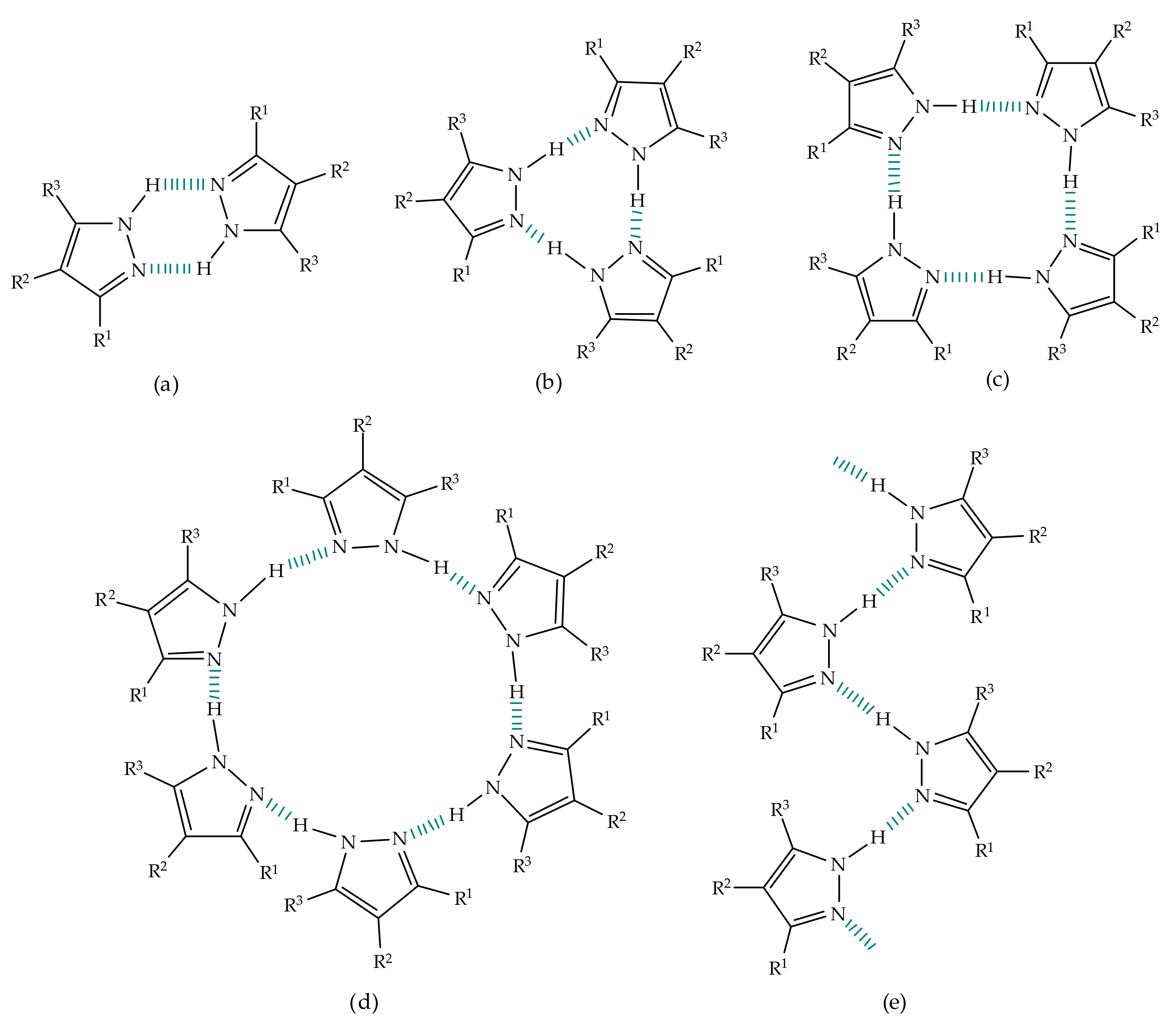
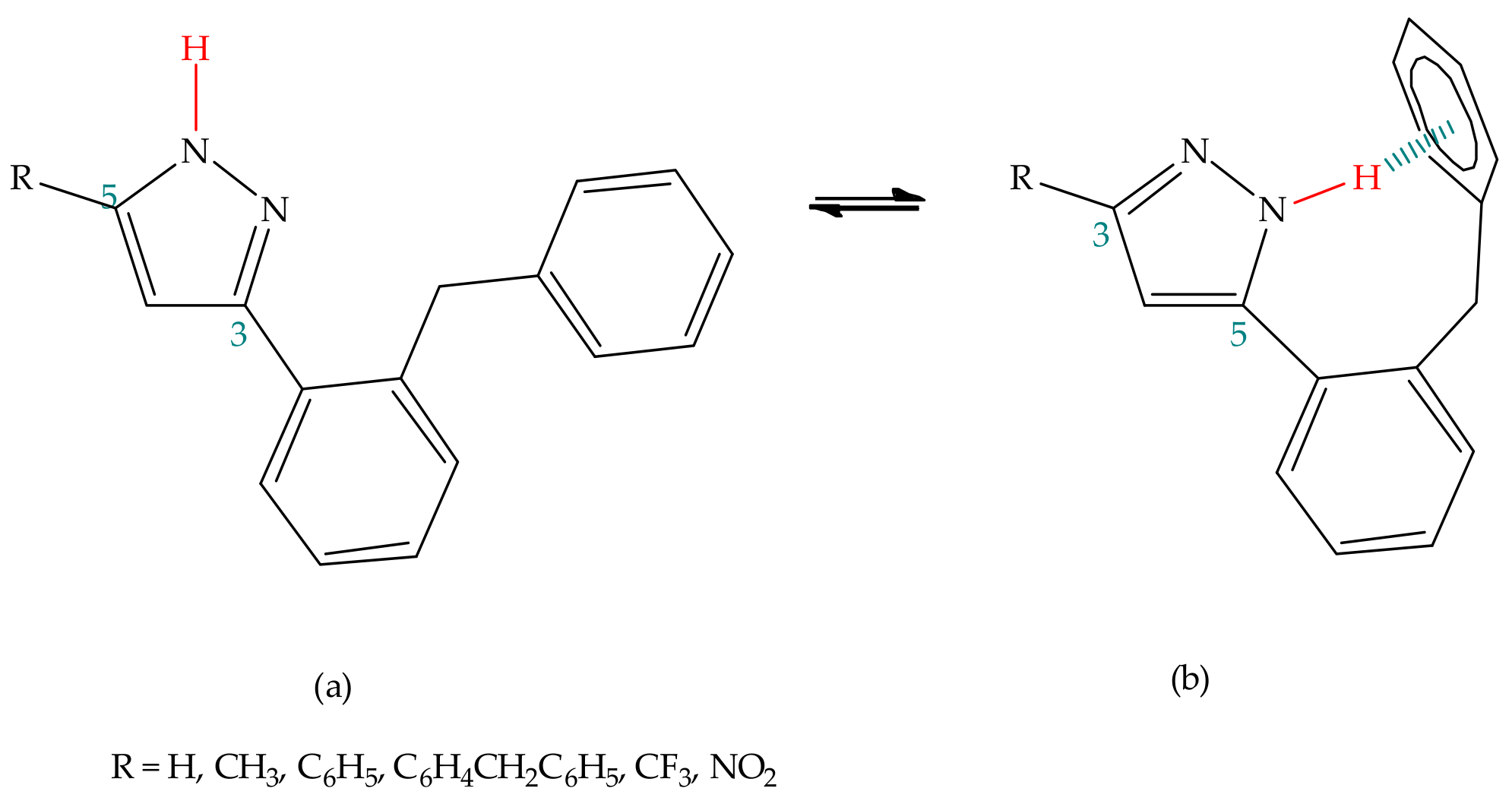
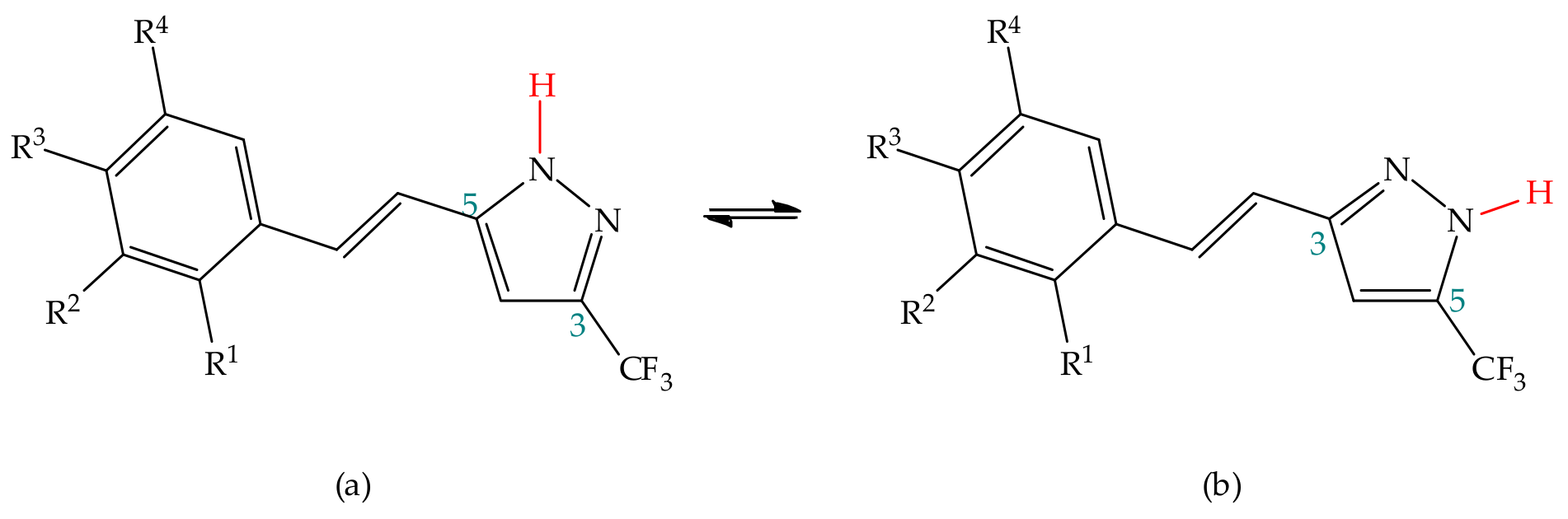
3.1.2. Experimental Studies
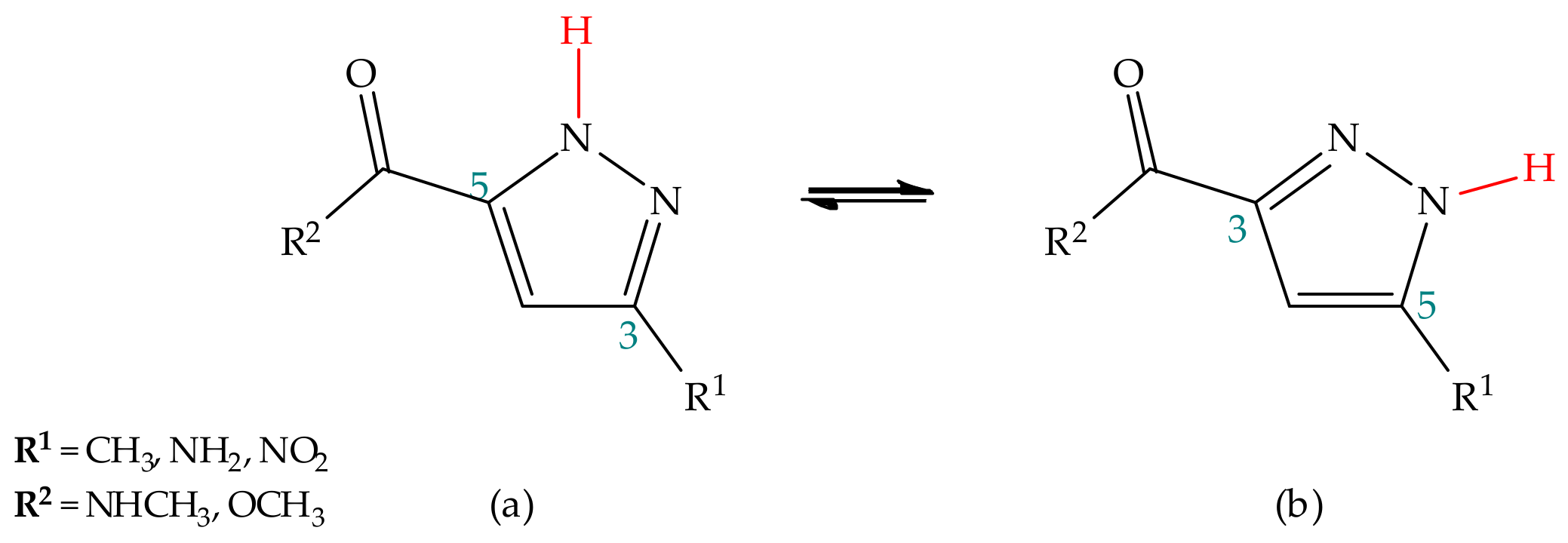
Substituent Effect
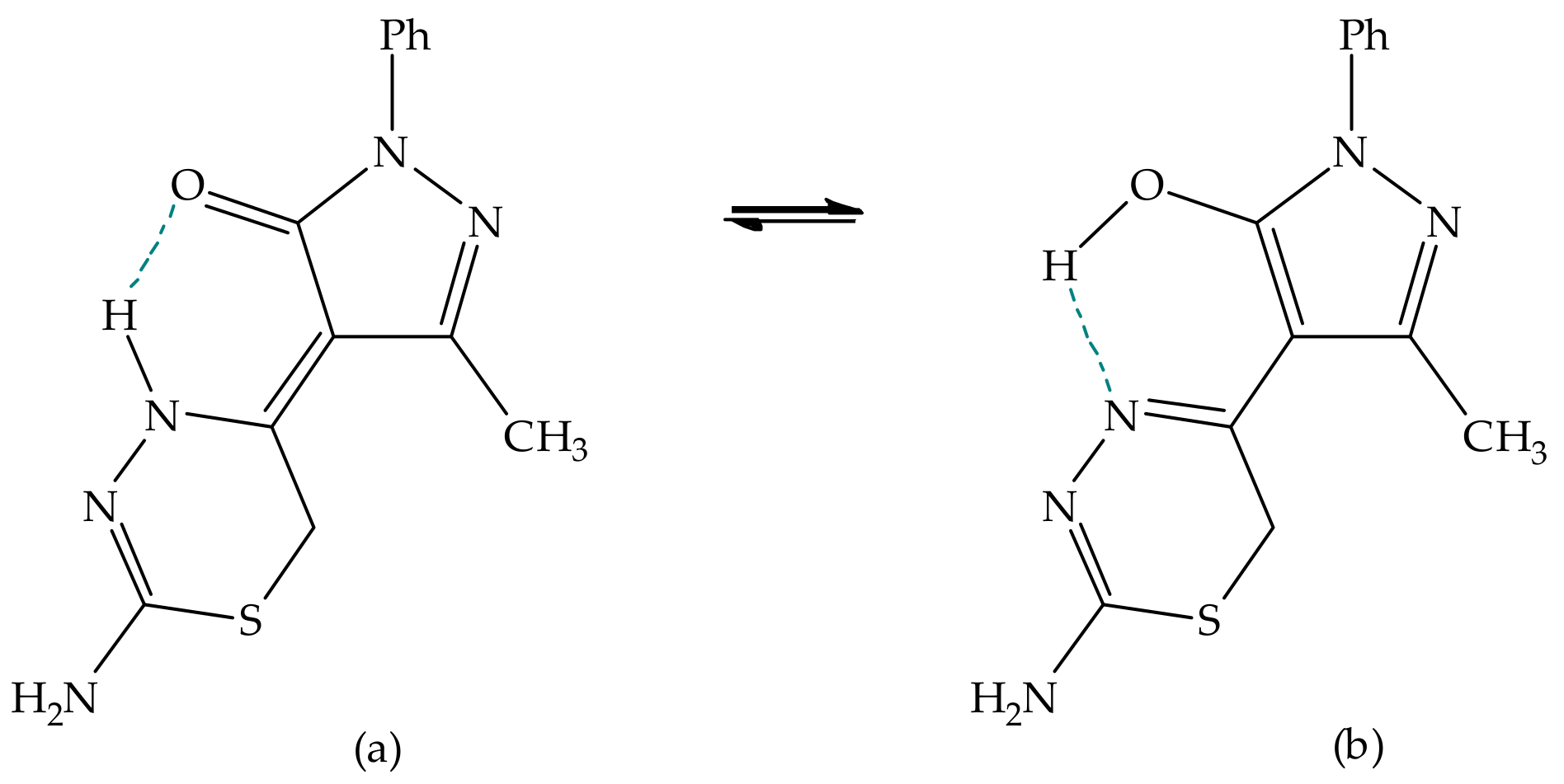
Environmental Effect
4. Pyrazoles in Organic Synthesis
5. 3(5)-Aminopyrazole
5.1. Tautomerism in 3(5)-Aminopyrazole
5.1.1. Theoretical Studies
5.1.2. Experimental Studies
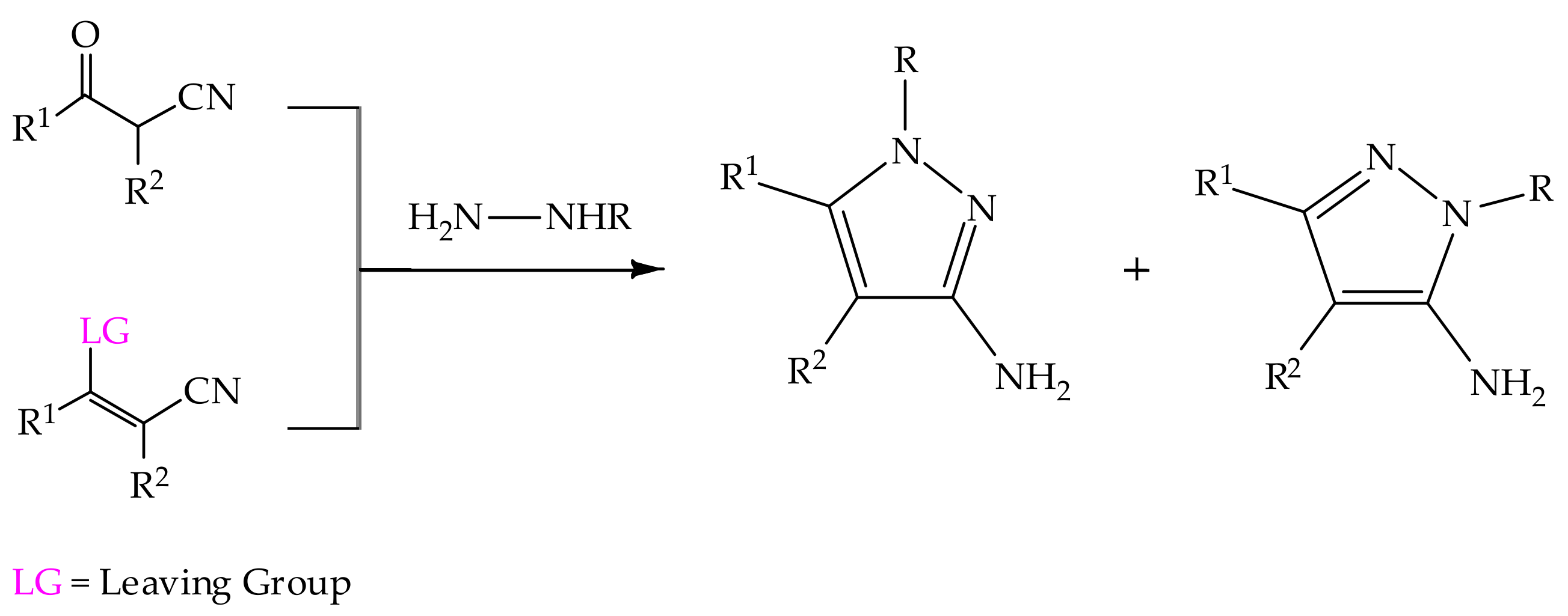
5.2. 3(5)-Aminopyrazole in Heterocyclic Synthesis
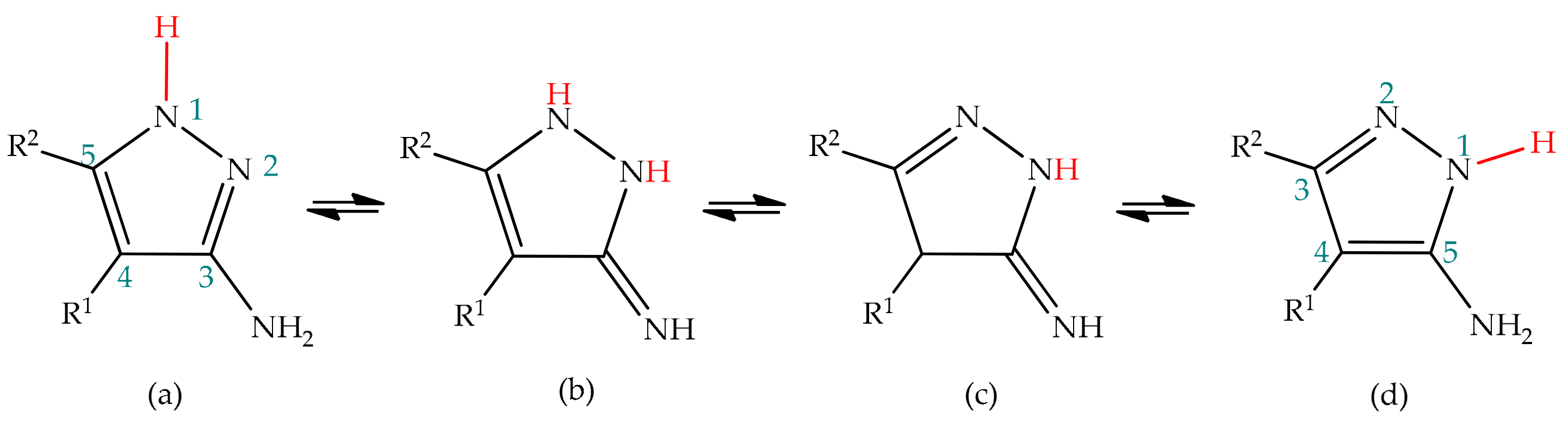
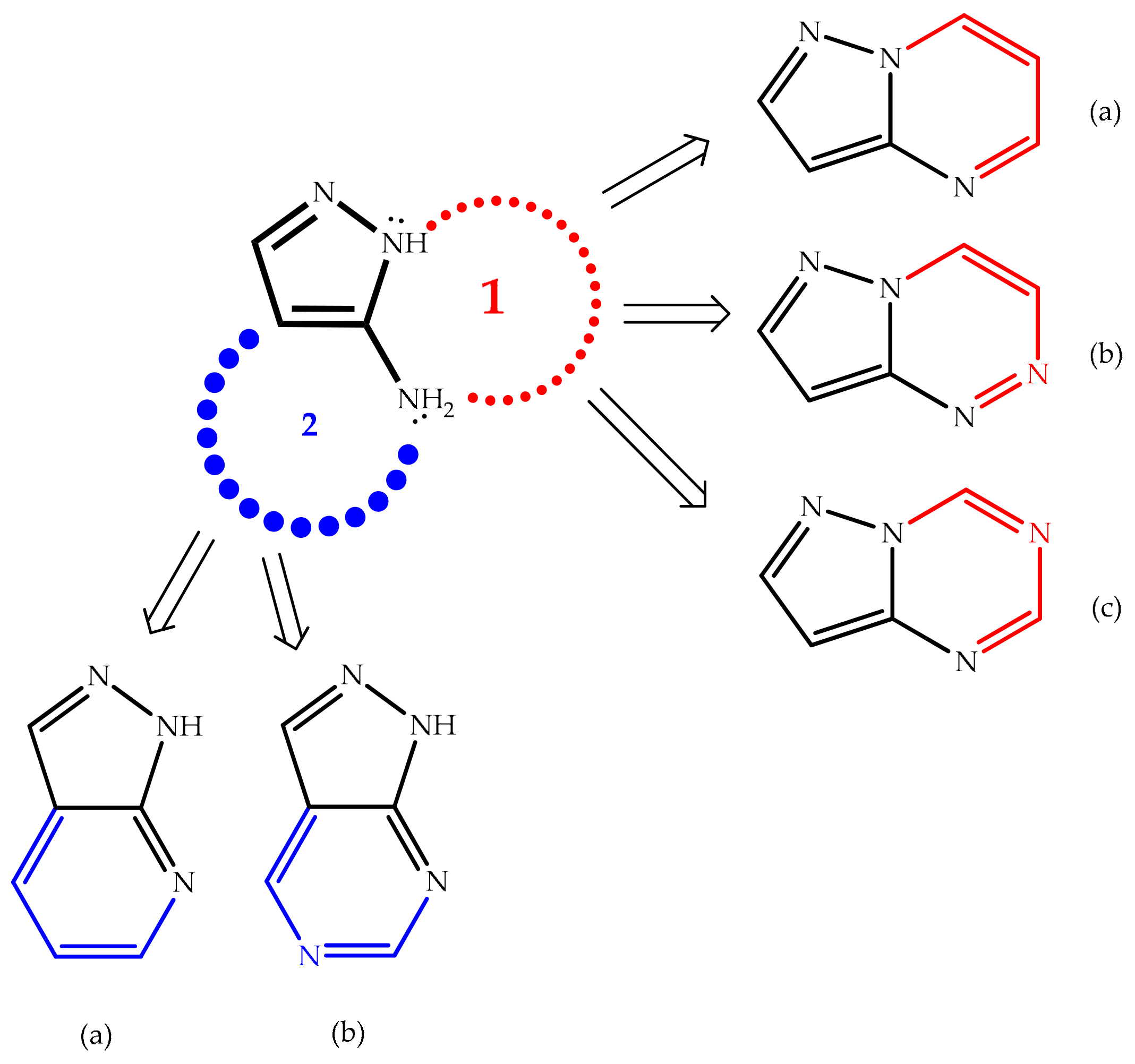
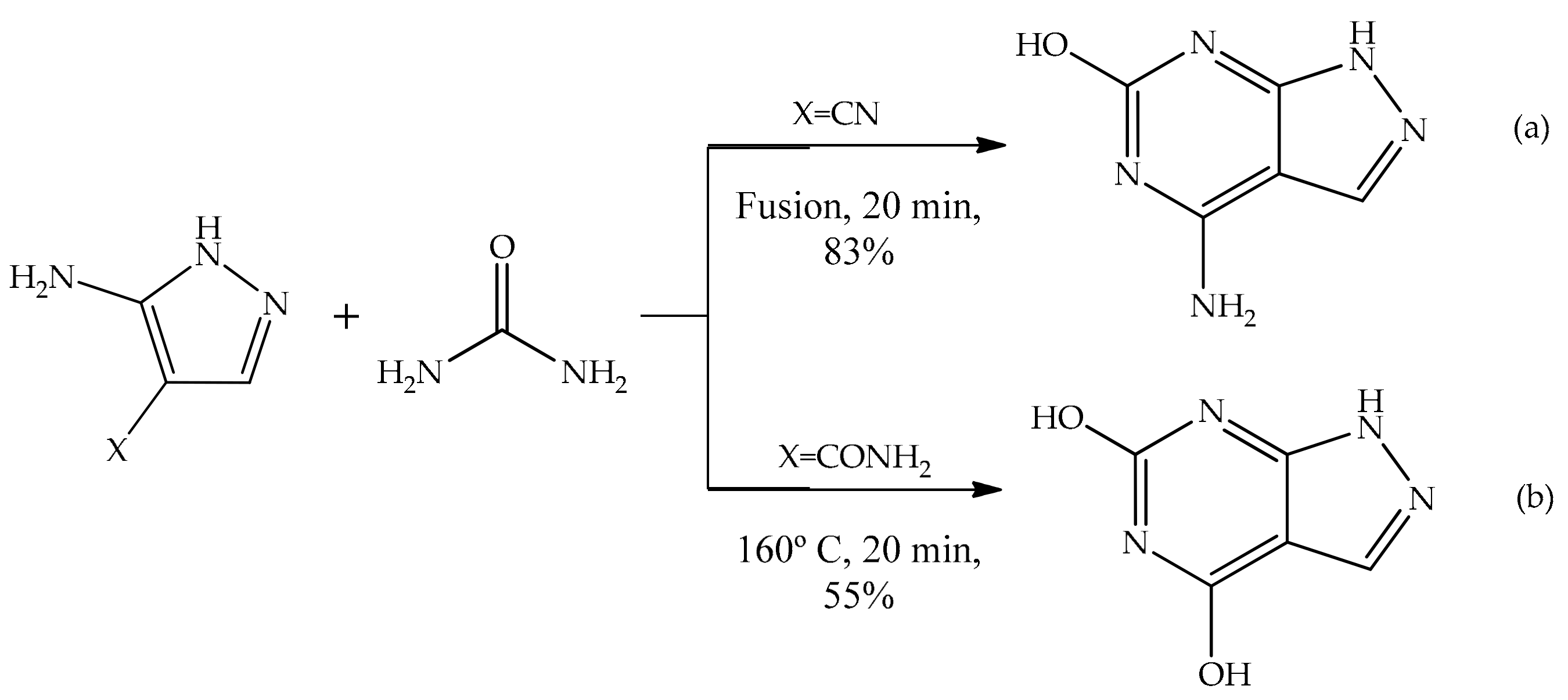
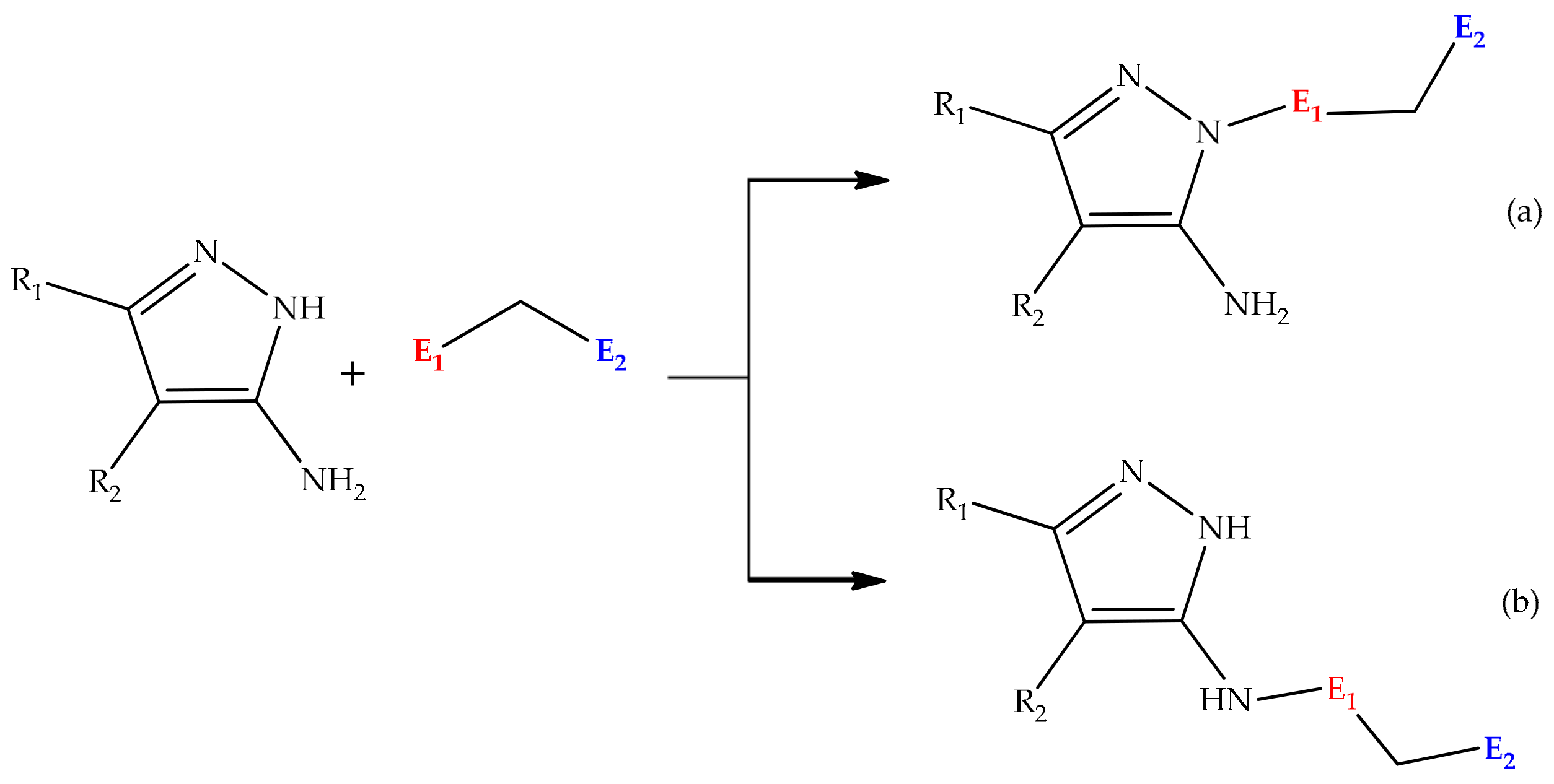
5.2.1. Intramolecular Cyclization of 3(5)-Aminopyrazole
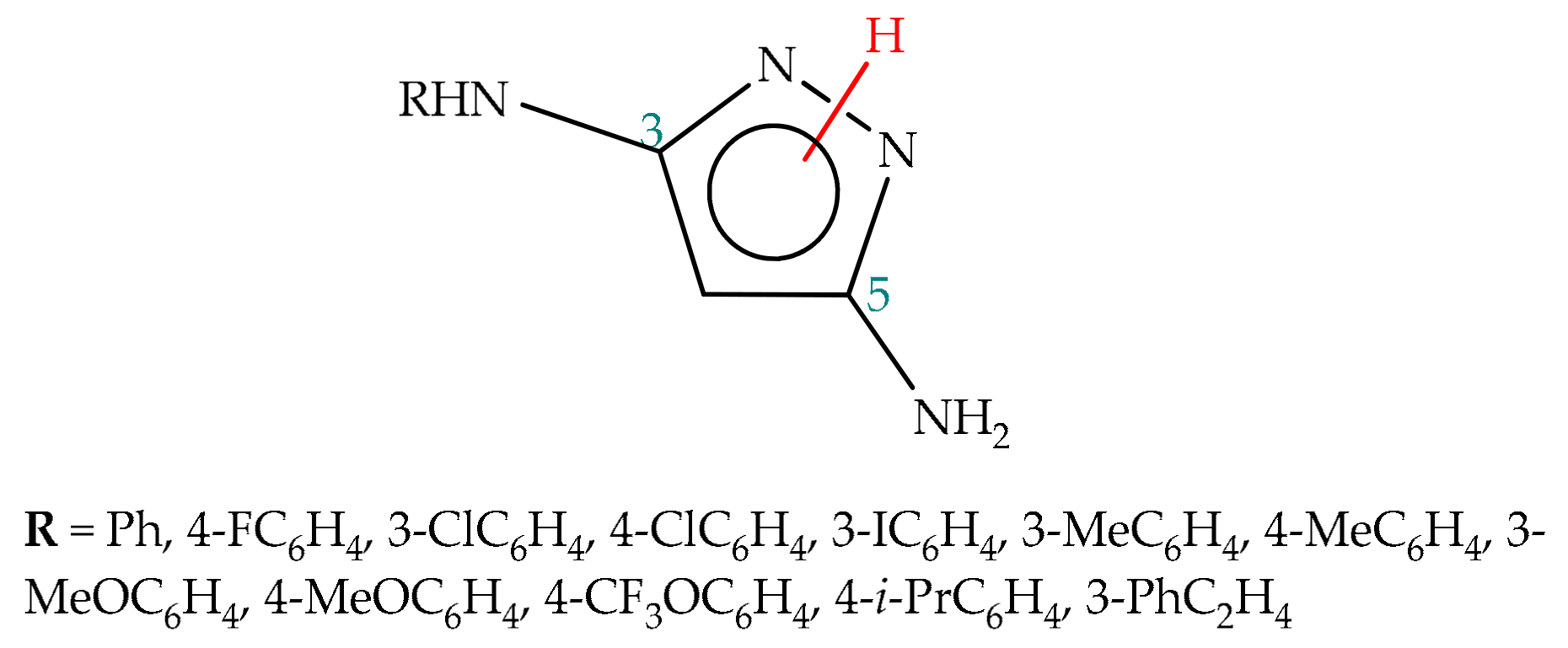
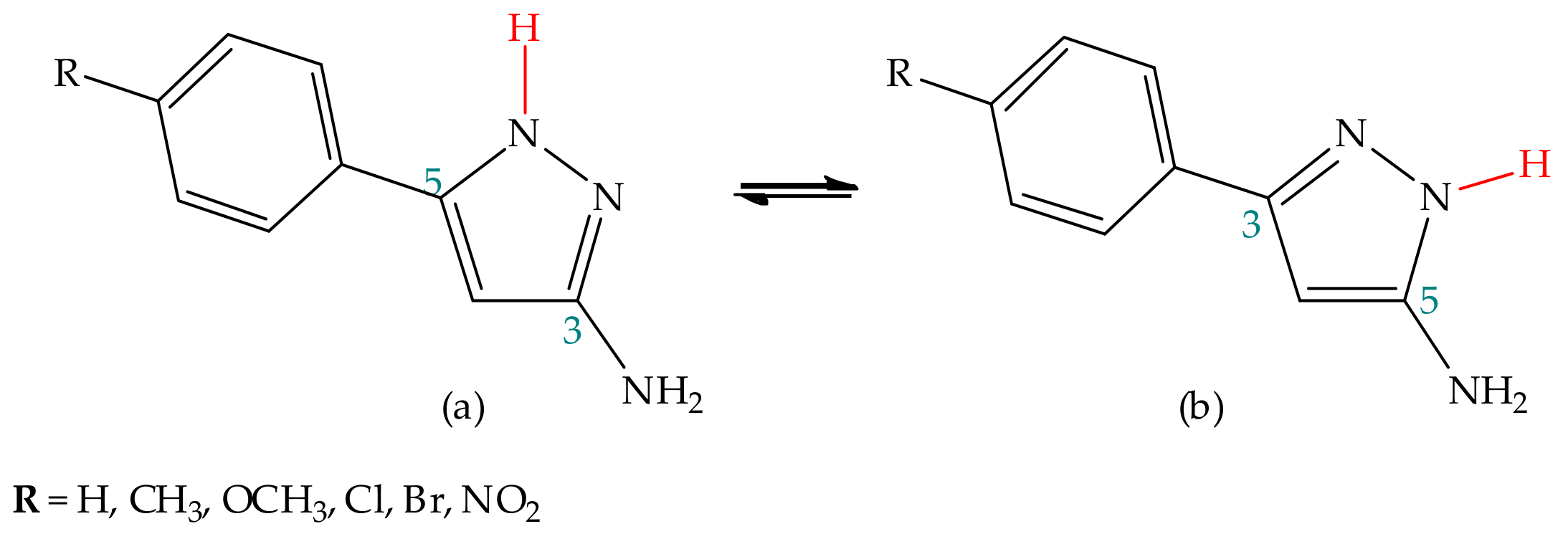
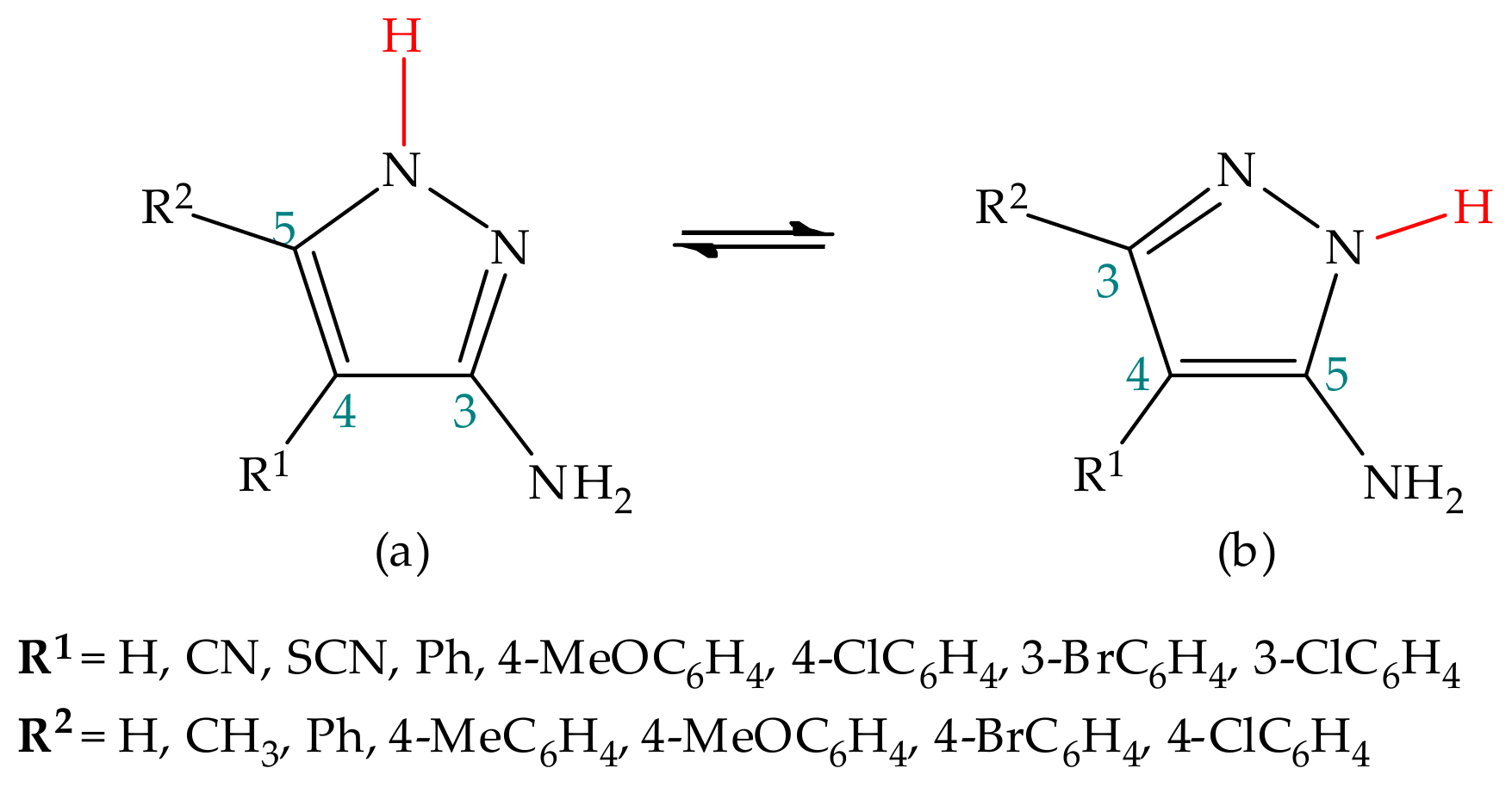
5.2.2. Substituent Effect
5.2.3. Environmental Effect
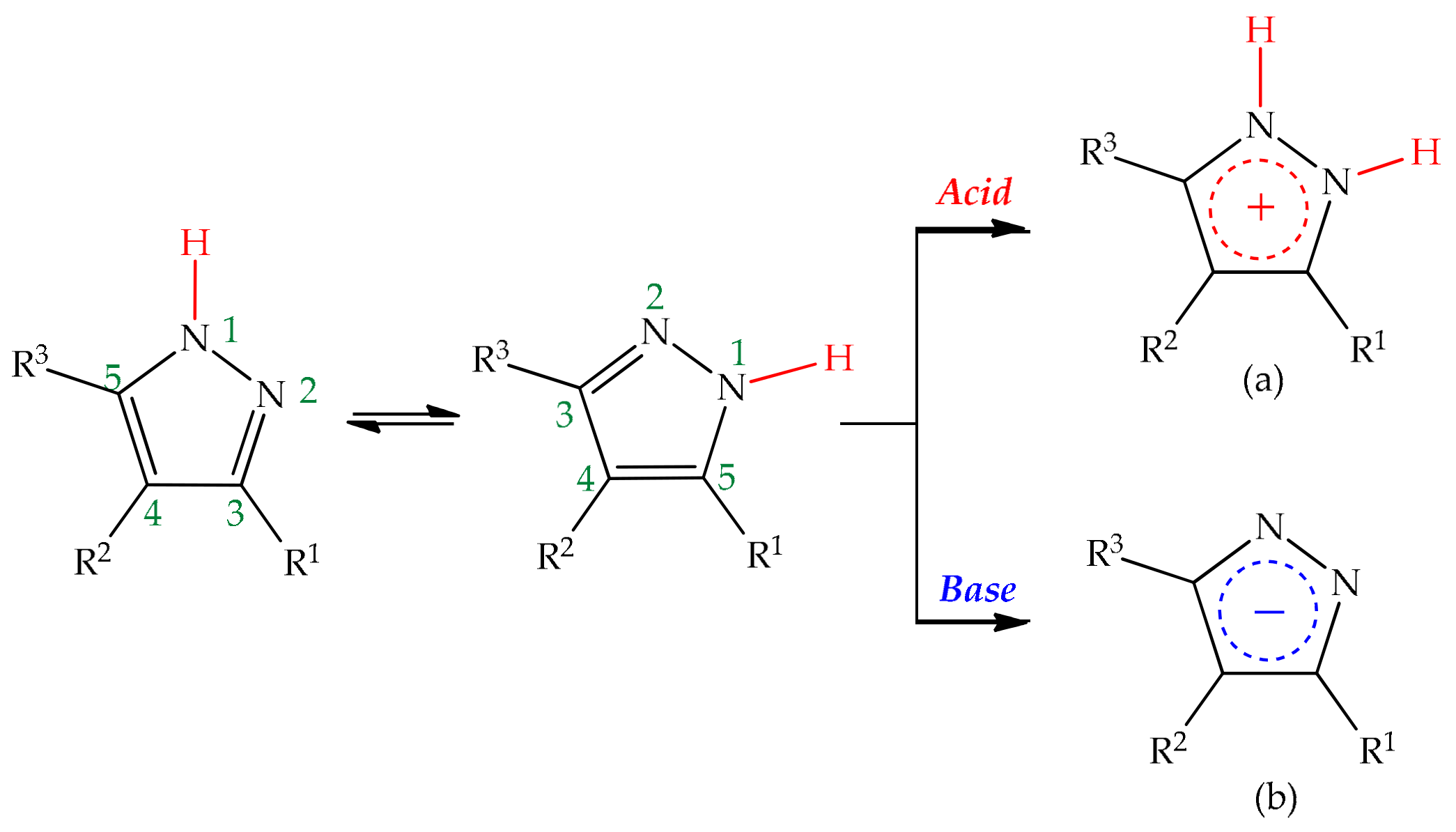
5.2.4. Acid Catalysis
5.2.5. Base Catalysis
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Schofield, K.; Grimmett, M.R.; Grimmett, M.R.; Keene, B.R. Heteroaromatic Nitrogen Compounds: The Azoles; Cambridge University Press: Cambridge, UK, 1976. [Google Scholar]
- Andes, D.R.; Dismukes, W.E. Azoles. In Essentials of Clinical Mycology, 2nd ed.; Kauffman, C.A., Pappas, P.G., Sobel, J.D., Dismukes, W.E., Eds.; Springer: New York, NY, USA, 2011; pp. 61–93. [Google Scholar]
- Kumar, S.S.; Kavitha, H.P. Synthesis and Biological Applications of Triazole Derivatives—A Review. Mini. Rev. Org. Chem. 2013, 10, 40–65. [Google Scholar] [CrossRef]
- Wei, C.X.; Bian, M.; Gong, G.H. Tetrazolium compounds: Synthesis and applications in medicine. Molecules 2015, 20, 5528–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, S.Q.; Young, D.J.; Hor, T.S.A. Nitrogen-rich azoles as ligand spacers in coordination polymers. Chem. Asian J. 2011, 6, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Ramsden, C.A.; Joule, J.A.; Zhdankin, V.V. Reactivity of Five-membered Rings with Two or More Heteroatoms. In Handbook of Heterocyclic Chemistry, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 473–604. [Google Scholar]
- Behr, L.C.; Fusco, R.; Jarboe, C.H. Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings; John Wiley & Sons: New York, NY, USA, 1967; p. 150. [Google Scholar]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Vicentini, C.B.; Mares, D.; Tartari, A.; Manfrini, M.; Forlani, G. Synthesis of Pyrazole Derivatives and Their Evaluation as Photosynthetic Electron Transport Inhibitors. J. Agric. Food Chem. 2004, 52, 1898–1906. [Google Scholar] [CrossRef]
- El-Mekabaty, A. Utility of 5-amino-1-phenyl-1H-pyrazole-4-carboxamide in heterocyclic synthesis. Synth. Commun. 2014, 44, 875–896. [Google Scholar] [CrossRef]
- Kumari, S.; Paliwal, S.; Chauhan, R. Synthesis of pyrazole derivatives possessing anticancer activity: Current status. Synth. Commun. 2014, 44, 1521–1578. [Google Scholar] [CrossRef]
- Katritzky, A.R. Advances in Heterocyclic Chemistry 76; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Shaabani, A.; Nazeri, M.T.; Afshari, R. 5-Amino-pyrazoles: Potent reagents in organic and medicinal synthesis. Mol. Divers. 2019, 23, 751–807. [Google Scholar] [CrossRef]
- Elmaati, T.M.A.; El-Taweel, F.M. New Trends In The Chemistry Of 5-Aminopyrazoles. J. Heterocycl. Chem. 2004, 41, 109–134. [Google Scholar] [CrossRef]
- Elnagdi, M.H.; Elmoghayar, M.R.H.; Elgemeie, G.E.H. Chemistry of Pyrazolopyrimidines. Adv. Heterocycl. Chem. 1987, 41, 319–376. [Google Scholar]
- Salem, M.A.; Helal, M.H.; Gouda, M.A.; EL-Gawad, H.H.A.; Shehab, M.A.M.; El-Khalafawy, A. Recent synthetic methodologies for pyrazolo[1,5-a]pyrimidine. Synth. Commun. 2019, 49, 1750–1776. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo [1, 5-a] pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.; Mahmoud, A.M. Believes versus evidence-based regio-orientation in the structure assignment of pyrazolo [1, 5-a] pyrimidines. J. Appl. Pharm. Sci. 2019, 9, 126–144. [Google Scholar]
- Brown, A.W. Recent Developments in the Chemistry of Pyrazoles, 1st ed.; Elsevier Inc.: San Diego, CA, USA, 2018; Volume 126. [Google Scholar]
- Anwar, H.F.; Elnagdi, M.H. Recent developments in aminopyrazole chemistry. ARKIVOC 2009, 2009, 198–250. [Google Scholar]
- Mert, S.; Kasimogullari, R.; Ok, S. A Short Review on Pyrazole Derivatives and their Applications. J. Postdr. Res. 2014, 2, 64–72. [Google Scholar]
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. [Google Scholar] [CrossRef]
- Kumar, H.; Saini, D.; Jain, S.; Jain, N. Pyrazole scaffold: A remarkable tool in the development of anticancer agents. Eur. J. Med. Chem. 2013, 70, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; Santos, M.S.d.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Kim, H.S.; Kurasawa, Y. Synthesis of pyrazoles and condensed pyrazoles. J. Heterocycl. Chem. 1999, 36, 321–332. [Google Scholar] [CrossRef]
- Schmidt, A.; Dreger, A. Recent Advances in the Chemistry of Pyrazoles. Properties, Biological Activities, and Syntheses. Curr. Org. Chem. 2011, 15, 1423–1463. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of Pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.J.; Alam, O.; Alam, P.; Naim, M.J. A Review on Pyrazole chemical entity and Biological Activity. Int. J. Pharma Sci. Res. 2015, 6, 1433–1442. [Google Scholar]
- Kumar, K.A.; Jayaroopa, P. Pyrazoles: Synthetic strategies and their pharmaceutical applications-an overview. Int. J. PharmTech Res. 2013, 5, 1473–1486. [Google Scholar]
- Chauhan, A.; Sharma, P.K.; Kaushik, N. Pyrazole: A versatile moiety. Int. J. ChemTech Res. 2011, 3, 11–17. [Google Scholar]
- Bekhit, A.A.; Hymete, A.; Bekhit, A.E.A.; Damtew, A.; Aboul-Enein, H.Y. Pyrazoles as Promising Scaffold for the Synthesis of Anti-Inflammatory and/or Antimicrobial Agent: A Review. Mini Rev. Med. Chem. 2012, 10, 1014–1033. [Google Scholar] [CrossRef] [PubMed]
- Nimbalkar, S.; Hote, S.V. Pyrazole Derivatives and their Synthesis—A review. Int. J. Recent Innov. Trends Comput. Commun. 2015, 3, 61–65. [Google Scholar]
- Fustero, S.; María, S.; Barrio, P.; Sim, A. From 2000 to Mid-2010 : A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Giller, S.A.; Mazheika, I.B.; Grandberg, I.I. Electron density distribution in heterocyclic systems with two vicinal nitrogen atoms. II. Pyrazole dipole moments. Khimiya Geterotsiklicheskikh Soedin. 1965, 1, 103–106. [Google Scholar] [CrossRef]
- Chermahini, A.N.; Teimouri, A.; Beni, A.S.; Dordahan, F. Theoretical studies on the effect of substituent in the proton transfer reaction of 4-substituted pyrazoles. Comput. Theor. Chem. 2013, 1008, 67–73. [Google Scholar] [CrossRef]
- Catalán, J.; Sánchez-Cabezudo, M.; de Paz, J.L.G.; Elguero, J.; Taft, R.W.; Anvia, F. The tautomerism of 1,2,3-triazole, 3(5)-methylpyrazole and their cations. J. Comput. Chem. 1989, 10, 426–433. [Google Scholar] [CrossRef]
- Catalan, J.; Abboud, J.L.M.; Elguero, J. Basicity and Acidity of Azoles. Adv. Heterocycl. Chem. 1987, 41, 187–274. [Google Scholar]
- Castaneda, J.P.; Denisov, G.S.; Kucherov, S.Y.; Schreiber, V.M.; Shurukhina, A.V. Infrared and ab initio studies of hydrogen bonding and proton transfer in the complexes formed by pyrazoles. J. Mol. Struct. 2003, 660, 25–40. [Google Scholar] [CrossRef]
- Claramunt, R.M.; López, C.; María, M.D.S.; Sanz, D.; Elguero, J. The use of NMR spectroscopy to study tautomerism. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 169–206. [Google Scholar] [CrossRef]
- Pérez, J.; Riera, L. Pyrazole complexes and supramolecular chemistry. Eur. J. Inorg. Chem. 2009, 33, 4913–4925. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Cornago, P.; Torres, V.; Pinilla, E.; Torres, M.R.; Samat, A.; Lokshin, V.; Valés, M.; Elguero, J. The structure of pyrazoles in the solid state: A combined CPMAS, NMR, and crystallographic study. J. Org. Chem. 2006, 71, 6881–6891. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Goya, P.; Elguero, J.; Singh, S.P. A simple approach to the tautomerism of aromatic heterocycles. Natl. Acad. Sci. Lett. 2007, 30, 139–159. [Google Scholar]
- Chermahini, A.N.; Teimouri, A. Theoretical studies on proton transfer reaction of 3(5)-substituted pyrazoles. J. Chem. Sci. 2014, 126, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Oziminski, W.P. The kinetics of water-assisted tautomeric 1,2-proton transfer in azoles: A computational approach. Struct. Chem. 2016, 27, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Knorr, L. Ueber Abkömmlinge der Phenolform des 1-Phenyl-3-methyl-5-pyrazolons. Eur. J. Inorg. Chem. 1895, 28, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Elattar, K.M.; Fadda, A.A. Chemistry of antipyrine. Synth. Commun. 2016, 46, 1567–1594. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Hall, C.D.; El-Gendy, B.E.D.M.; Draghici, B. Tautomerism in drug discovery. J. Comput. Aided. Mol. Des. 2010, 24, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; van der Zwan, G.; Antonov, L. Tautomerism: Introduction, History, and Recent Developments in Experimental and Theoretical Methods. In Tautomerism: Methods and Theories, 1st ed.; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 1–24. [Google Scholar]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to Pi-electron delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef] [PubMed]
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. Tautomerism of Heterocycles: Five-Membered Rings with Two or More Heteroatoms. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 159–323. [Google Scholar]
- Minkin, V.I.; Olekhnovich, L.P.; Zhdanov, Y.A. Molecular Design of Tautomeric Compounds; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1981; Volume 14. [Google Scholar]
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 2–84. [Google Scholar]
- Martin, Y.C. Let’s not forget tautomers. J. Comput. Aided. Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkorta, I.; Elguero, J. 1,2-Proton shifts in pyrazole and related systems: A computational study of [1,5]-sigmatropic migrations of hydrogen and related phenomena. J. Chem. Soc. Perkin Trans. 1998, 2, 2497–2504. [Google Scholar] [CrossRef]
- Favre, H.A.; Powell, W.H. Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Katritzky, A.R.; Karelson, M.; Harris, P.A. Prototropic Tautomerism of Heteroaromatic Compounds. Heterocycles 1991, 32, 329–369. [Google Scholar] [CrossRef]
- Nagy, P.I. Theoretical Consideration of In-Solution Tautomeric Equilibria in Relation to Drug Design. Tautomerism 2016, 2016, 113–146. [Google Scholar]
- Clark, T. A Handbook of Computational Chemistry: A Practical Guide to Chemical Structure and Energy Calculations; J. Wiley: New York, NY, USA, 1985. [Google Scholar]
- Martin, Y.C. Experimental and pKa prediction aspects of tautomerism of drug-like molecules. Drug Discov. Today Technol. 2018, 27, 59–64. [Google Scholar] [CrossRef]
- Larina, L.I. Tautomerism and Structure of Azoles: Nuclear Magnetic Resonance Spectroscopy. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier Ltd.: San Diego, CA, USA, 2018; Volume 126, pp. 233–321. [Google Scholar]
- Szajda, M.; Lam, J.N. Thiophenes and their Benzo Derivatives: Structure. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Pergamon Press: Oxford, UK, 1996; Volume 2, pp. 491–605. [Google Scholar]
- Simons, M.; Topper, A.; Sutherland, B.; Seybold, P.G. A Class Project Combining Organic Chemistry, Quantum Chemistry, and Statistics. Annu. Rep. Comput. Chem. 2011, 7, 237–249. [Google Scholar]
- Lin, H.; Wu, D.; Liu, L.; Jia, D. Theoretical study on molecular structures, intramolecular proton transfer reaction, and solvent effects of 1-phenyl-3-methyl-4-(6-hydro-4-amino-5-sulfo-2,3-pyrazine)-pyrazole-5-on e. J. Mol. Struct. THEOCHEM 2008, 850, 32–37. [Google Scholar] [CrossRef]
- Catalán, J.; Menéndez, M.; Laynez, J.; Claramunt, R.M.; Bruix, M.; De Mendoza, J.; Elguero, J. Basicity of azoles. Basicity of C-Aminopyrazoles in Relation to Tautomeric and Protonation Studies. J. Heterocycl. Chem. 1985, 22, 997–1000. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Liebman, J.F. The annular tautomerism of imidazoles and pyrazoles: The possible existence of nonaromatic forms. Struct. Chem. 2006, 17, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Jarończyk, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struct. THEOCHEM 2004, 673, 17–28. [Google Scholar] [CrossRef]
- Cornago, P.; Claramunt, R.M.; Bouissane, L.; Elguero, J. A molecular balance to measure the strength of N-H π hydrogen bonds based on the tautomeric equilibria of C-benzylphenyl substituted NH-pyrazoles. Tetrahedron 2008, 64, 3667–3673. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. A theoretical study on the tautomerism of C-carboxylic and methoxycarbonyl substituted azoles. Struct. Chem. 2005, 16, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Blanco, F.; Elguero, J. A theoretical comparison of the chemical shifts of three related heterocycles: 1H-pyrazoles, 1H-1,2,4-triazoles and 1H-1,2,4-diazaphospholes. J. Mol. Struct. THEOCHEM 2010, 942, 1–6. [Google Scholar] [CrossRef]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Nieto, C.I.; Cabildo, P.C.; Cornago, P.M.; Sanz, D.; Claramunt, R.M.; Alkorta, I.; Elguero, J.; García, J.A.; López, A.; Acuña-Castroviejo, D. Synthesis, structure and biological activity of 3(5)-trifluoromethyl-1H-pyrazoles derived from hemicurcuminoids. J. Mol. Struct. 2015, 1100, 518–529. [Google Scholar] [CrossRef]
- Kusakiewicz-Dawid, A.; Porada, M.; Dziuk, B.; Siodlak, D. Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules 2019, 24, 2632. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Blanco, F.; Elguero, J. Proton transfer in C-halogen pyrazole cyclamers. A theoretical study. Struct. Chem. 2008, 19, 191–198. [Google Scholar] [CrossRef]
- de Paz, G.L.; Elguero, J.; Foces-Foces, C.; Llamas-Saiz, A.L.; Aguilar-Parrilla, F.; Kleind, O.; Limbach, H.-H. Theoretical study of the structure and tautomerism of N1-unsubstituted pyrazoles in the solid state. J. Chem. Soc. Perkin Trans. 1997, 5, 101–109. [Google Scholar] [CrossRef]
- Horta, P.; Secrieru, A.; Coninckx, A.; Cristiano, M.S. Quinolones for Applications in Medicinal Chemistry: Synthesis and Structure. In Targets in Heterocyclic Systems; Attanasi, O., Merino, P., Spinelli, D., Eds.; Società Chimica Italiana: Rome, Italy, 2018; pp. 260–297. [Google Scholar]
- Horta, P. Quinolone-Hydroxyquinoline Tautomerism in Quinolone 3-Esters. Preserving the 4-Oxoquinoline Structure To Retain Antimalarial Activity. J. Org. Chem. 2015, 80, 12244–12257. [Google Scholar] [CrossRef] [PubMed]
- Horta, P.; Henriques, M.S.C.; Brás, E.M.; Murtinheira, F.; Nogueira, F.; O’Neill, P.M.; Paixaõ, J.A.; Fausto, R.; Cristiano, M.L.S. On the ordeal of quinolone preparation via cyclisation of aryl-enamines; Synthesis and structure of ethyl 6-methyl-7-iodo-4-(3-iodo-4-methylphenoxy)-quinoline-3-carboxylate. Pure Appl. Chem. 2017, 89, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.I.; Tejada, F.R.; Messer, W.S. Theoretical studies of the tautomeric equilibria for five-member N-heterocycles in the gas phase and in solution. J. Phys. Chem. B 2005, 109, 22588–22602. [Google Scholar] [CrossRef] [PubMed]
- Beni, A.S.; Chermahini, Z.J. Theoretical studies on tautomerism of 1H-pyrazole-5-thiol. Struct. Chem. 2013, 24, 1713–1723. [Google Scholar] [CrossRef]
- Zerbi, G. Infrared Spectra of Pyrazoles-I Pyrazoles Mono-Alkyl Substituted. Spectrochim. Acta 1962, 18, 407–423. [Google Scholar]
- Billes, F.; Endrédi, H.; Jalsovszky, G. Vibrational spectroscopy of diazoles. J. Mol. Struct. THEOCHEM 1999, 465, 157–172. [Google Scholar] [CrossRef]
- Maquestiau, A.; Flammang, R. Studies of prototropy by mass spectrometric methods. Mass Spectrom. Rev. 1982, 1, 237–255. [Google Scholar] [CrossRef]
- Huang, A.; Wo, K.; Lee, S.Y.C.; Kneitschel, N.; Chang, J.; Zhu, K.; Mello, T.; Bancroft, L.; Norman, N.J.; Zheng, S.L. Regioselective Synthesis, NMR, and Crystallographic Analysis of N1-Substituted Pyrazoles. J. Org. Chem. 2017, 82, 8864–8872. [Google Scholar] [CrossRef]
- Puello, J.Q. Structure and tautomerism of 3(5)-amino-5(3)-arylpyrazoles in the solid state and in solution: An X-ray and NMR study. Tetrahedron 1997, 53, 10783–10802. [Google Scholar] [CrossRef]
- Kruszyk, M.; Jessing, M.; Kristensen, J.L.; Jørgensen, M. Computational Methods to Predict the Regioselectivity of Electrophilic Aromatic Substitution Reactions of Heteroaromatic Systems. J. Org. Chem. 2016, 81, 5128–5134. [Google Scholar] [CrossRef]
- Claramunt, R.M. Substituent effects on the 15N NMR Parameters of Azoles. Magn. Reson. Chem. 2017, 35, 35–75. [Google Scholar] [CrossRef]
- Faure, R.; Vincent, É.-J.; Rousseau, A.; Claramunt, R.M.; Elguero, J. High resolution 13C nuclear magnetic resonance spectra of solid pyrazoles. Application to annular tautomerism. Can. J. Chem. 1988, 66, 1141–1146. [Google Scholar] [CrossRef]
- Elguero, J.; Marzin, C.; Roberts, J.D. Carbon-13 Magnetic Resonance Studies of Azoles. Tautomerism, Shift Reagent Effects, and Solvent Effects. J. Org. Chem. 1974, 39, 357–363. [Google Scholar] [CrossRef]
- Elguero, J.; Fruchier, A.; Pellegrin, V. Annular tautomerism in the solid state: A high resolution n.m.r. study. J. Chem. Soc. Chem. Commun. 1981, 23, 1207–1208. [Google Scholar] [CrossRef]
- López-Cara, L.C.; Carrión, M.D.; Camacho, M.E.; Gallo, M.A.; Espinosa, A.; Choquesillo-Lazarte, D.; Gonzalez-Pérez, J.M.; Entrena, G.A. 1H, 13C NMR, X-ray and conformational studies of new 1-alkyl-3-benzoyl-pyrazole and 1-alkyl-3-benzoyl-pyrazoline derivatives. Magn. Reson. Chem. 2008, 46, 878–885. [Google Scholar] [CrossRef]
- García, M.Á.; Claramunt, R.M.; Elguero, J. 13 C and 15 N NMR chemical shifts of 1-(2′,4′-dinitrophenyl) and 1-(2′,4′,6′- trinitrophenyl) pyrazoles in the solid state and in solution. Magn. Reson. Chem. 2008, 46, 697–700. [Google Scholar] [CrossRef]
- Klein, O.; Aguilar-Parrilla, F.; Lopez, J.M.; Jagerovic, N.; Elguero, J.; Limbach, H.H. Dynamic NMR study of the mechanisms of double, triple, and quadruple proton and deuteron transfer in cyclic hydrogen bonded solids of pyrazole derivatives. J. Am. Chem. Soc. 2004, 126, 11718–11732. [Google Scholar] [CrossRef] [Green Version]
- Begtrup, M.; Boyer, G.; Cabildo, P.; Cativiela, C.; Claramunt, R.M.; Elguero, J.; García, J.I.; Toiron, C.; Vedsø, P. 13C NMR of pyrazoles. Magn. Reson. Chem. 1993, 31, 107–168. [Google Scholar] [CrossRef]
- Chenon, M.T.; Coupry, C.; Grant, D.M.; Pugmire, R.J. Carbon-13 Magnetic Resonance Study of Solvent Stabilized Tautomerism in Pyrazoles. J. Org. Chem. 1977, 42, 659–661. [Google Scholar] [CrossRef]
- Kalikhman, I.D.; Lopyrev, V.A.; Shibanova, E.F.; Pestunovich, V.A.; Voronkov, M.G. Concentration dependence of the rate of prototropic rearrangement of azoles in hexamethylphosphoric triamide. Russ. Chem. Bull. 1977, 26, 2223. [Google Scholar] [CrossRef]
- Cabildo, P.; Claramunt, R.M.; Elguero, J. 13C NMR chemical shifts of N-unsubstituted- and N-methyl-pyrazole derivatives. Org. Magn. Reson. 1984, 22, 603–607. [Google Scholar] [CrossRef]
- Lopez, C.; Claramunt, R.M.; Trofimenko, S.; Elguero, J. A 13 C NMR spectroscopy study of the structure of N-H pyrazoles and indazoles. Can. J. Chem. 1993, 71, 678–684. [Google Scholar] [CrossRef]
- Bruix, M.; de Mendoza, J.; Claramunt, R.M.; Elguero, J. NMR Studies in the Heterocyclic Series. 27*—Carbon-13 NMR determination of the protonation site of aminopyrazoles in trifluoroacetic acid. Magn. Reson. Chem. 1985, 23, 367–374. [Google Scholar] [CrossRef]
- Provasi, P.; Jimeno, M.L.; Alkorta, I.; Reviriego, F.; Elguero, J.; Jokisaari, J. The 1H NMR spectrum of pyrazole in a nematic phase. Magn. Reson. Chem. 2016, 54, 637–640. [Google Scholar] [CrossRef]
- Tensmeyer, L.G.; Ainsworth, C. Proton Magnetic Resonance Studies of Pyrazoles. J. Org. Chem. 1966, 311, 1878–1883. [Google Scholar] [CrossRef]
- Baldy, A.; Eleuero, J.; Faure, R.; Pierrot, M.; Vincent, E.J. Dynamic Intermolecular Tautomerism of 3, 5-Dimethylpyrazole in the Solid State by 13C CP/MAS NMR Spectroscopy and X-ray Crystallography. J. Am. Chem. Soc. 1985, 107, 5290–5291. [Google Scholar] [CrossRef]
- Foces-Foces, C.; Echevarría, A.; Jagerovic, N.; Alkorta, I.; Elguero, J.; Langer, U.; Klein, O.; Minguet-Bonvehí, M.; Limbach, H.H. A solid-state NMR, X-ray diffraction, and ab initio computational study of hydrogen-bond structure and dynamics of pyrazole-4-carboxylic acid chains. J. Am. Chem. Soc. 2001, 123, 7898–7906. [Google Scholar] [CrossRef]
- Hall, D.G. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; WILEY-VCH: Weinheim, Germany, 2005. [Google Scholar]
- McLaughlin, M.; Marcantonio, K.; Chen, G.Y.; Davies, I.W. A simple, modular method for the synthesis of 3,4,5-trisubstituted pyrazoles. J. Org. Chem. 2008, 73, 4309–4312. [Google Scholar] [CrossRef]
- Kost, A.N.; Grandberg, I.I. Progress in Pyrazole Chemistry. Adv. Heterocycl. Chem. 1966, 6, 347–429. [Google Scholar]
- Halcrow, M.A. Pyrazoles and pyrazolides—Flexible synthons in self-assembly. J. Chem. Soc. Dalt. Trans. 2009, 9226, 2059–2073. [Google Scholar] [CrossRef]
- Aggarwal, R.; Kumar, S. 5-Aminopyrazole as precursor in design and synthesis of fused pyrazoloazines. Beilstein J. Org. Chem. 2018, 14, 203–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Paliwal, D.; Saini, D.; Thakur, A.; Aggarwal, S.; Kaushik, D. A comprehensive review on synthetic approach for antimalarial agents. Eur. J. Med. Chem. 2014, 85, 147–178. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Faure, R.; Vincent, E.-J.; Espada, M.; Elguero, J. Etude en Résonance Magnétique Nucléaire du Carbone-13 de Quelques Aminopyrazoles. Sur le Problème de la Détermination des Constantes d’Equilibre Tautomère. Org. Magn. Reson. 1979, 12, 587–592. [Google Scholar] [CrossRef]
- Dorn, H. Tautomerie und Nomenklatur der ‘Pyrazolone’ und Amino-pyrazole. J. Für Prakt. Chem. 1973, 315, 382–418. [Google Scholar] [CrossRef]
- Lim, F.P.L.; Tan, K.C.; Luna, G.; Tiekink, E.R.T.; Dolzhenko, A.V. A new practical synthesis of 3-amino-substituted 5-aminopyrazoles and their tautomerism. Tetrahedron 2019, 75, 2314–2321. [Google Scholar] [CrossRef]
- Abboud, J.L.M. Basicity of C-Substituted Pyrazoles in the Gas Phase: An Experimental (ICR) and Theoretical Study. J. Org. Chem. 1992, 57, 3938–3946. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Filyukov, D.V. Structure and tautomerism of 4-substituted 3(5)-aminopyrazoles in solution and in the solid state: NMR study and Ab initio calculations. Russ. J. Org. Chem. 2014, 50, 412–421. [Google Scholar] [CrossRef]
- Fichez, J.; Busca, P.; Prestat, G. Recent advances in aminopyrazoles synthesis and functionalization. Targets Heterocycl. Syst. 2017, 21, 322–347. [Google Scholar]
- El-Sattar, N.E.A.A.; Badawy, E.H.K.; Abdel-Mottaleb, M.S.A. Synthesis of Some Pyrimidine, Pyrazole, and Pyridine Derivatives and Their Reactivity Descriptors. J. Chem. 2018, 1–11. [Google Scholar] [CrossRef]
- Trujillo, C.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J. Computational Study of Proton Transfer in Tautomers of 3- and 5-Hydroxypyrazole Assisted by Water. ChemPhysChem 2015, 16, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Kolosov, M.A.; Beloborodov, D.A.; Orlov, V.D.; Dotsenko, V.V. Catalyst-free Biginelli-type synthesis of new functionalized 4,7-dihydropyrazolo[1,5-: A] pyrimidines. New J. Chem. 2016, 40, 7573–7579. [Google Scholar] [CrossRef] [Green Version]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically driven synthesis of pyrazolo[3,4- D ]pyrimidines as protein kinase inhibitors: An old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef] [PubMed]
- Murlykina, M.V.; Morozova, A.D.; Zviagin, I.M.; Sakhno, Y.I.; Desenko, S.M.; Chebanov, V.A. Aminoazole-based diversity-oriented synthesis of heterocycle. Front. Chem. 2018, 6, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.C.; Portilla, J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar]
- Kaur, N. Greener and Expeditious Synthesis of Fused Six-Membered N,N-Heterocycles Using Microwave Irradiation. Synth. Commun. 2015, 45, 1493–1519. [Google Scholar] [CrossRef]
- Charris-Molina, A.; Castillo, J.C.; Macías, M.; Portilla, J. One-Step Synthesis of Fully Functionalized Pyrazolo[3,4-b]pyridines via Isobenzofuranone Ring Opening. J. Org. Chem. 2017, 82, 12674–12681. [Google Scholar] [CrossRef] [PubMed]
- Fraley, M.E.; Hoffman, W.F.; Rubino, R.S.; Hungate, R.W.; Tebben, A.J.; Rutledge, R.Z.; McFall, R.C.; Huckle, W.R.; Kendall, R.L.; Coll, K.E.; et al. Synthesis and initial SAR studies of 3,6-disubstituted pyrazolo[1,5-a]pyrimidines: A new class of KDR kinase inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 2767–2770. [Google Scholar] [CrossRef]
- Almansa, C. Synthesis and SAR of a new series of COX-2-selective inhibitors: Pyrazolo[1,5-α]pyrimidines. J. Med. Chem. 2001, 44, 350–361. [Google Scholar] [CrossRef]
- Tsai, P.C.; Wang, I.J. Synthesis and solvatochromic properties of 3,6-bis-hetarylazo dyes derived from pyrazolo[1,5-a]pyrimidine. Dye. Pigment. 2008, 76, 575–581. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Hamdy, N.A.; Farag, A.M.; Fakhr, I.M.I. Synthesis of Some Novel Pyrazolo[1,5-a]pyrimidine, 1,2,4- Triazolo[1,5-a]pyrimidine, Pyrido[2,3-d]pyrimidine, Pyrazolo[5,1-c]-1,2,4-triazine and 1,2,4-Triazolo[5,1-c]-1,2,4- triazine Derivatives Incorporating a Thiazolo[3,2-a]benzimidazole Moiety. J. Heterocycl. Chem. 2008, 45, 1033–1037. [Google Scholar] [CrossRef]
- Zheng, N.; Pan, J.; Hao, Q.; Li, Y.; Zhou, W. Design, synthesis and biological evaluation of novel 3-substituted pyrazolopyrimidine derivatives as potent Bruton’s tyrosine kinase (BTK) inhibitors. Bioorg. Med. Chem. 2018, 26, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Michon, V.; Penhoat, C.H.d.; Tombret, F.; Gillardin, J.M.; Lepage, F.; Berthon, L. Preparation, structural analysis and anticonvulsant activity of 3- and 5-aminopyrazole N-benzoyl derivatives. Eur. J. Med. Chem. 1995, 30, 147–155. [Google Scholar] [CrossRef]
- Al-Qalaf, F.; Mandani, F.; Abdelkhalik, M.M.; Bassam, A.A. Synthesis of 5-substituted 3-amino-1H-pyrazole-4-carbonitriles as precursors for microwave assisted regiospecific syntheses of pyrazolo[1,5-a]pyrimidines. Molecules 2009, 14, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.E.M.; Elgemeie, G.H. Design, synthesis, docking, and antimicrobial evaluation of some novel pyrazolo[1,5-a] pyrimidines and their corresponding cycloalkane ring-fused derivatives as purine analogs. Drug Des. Devel. Ther. 2018, 12, 1785–1798. [Google Scholar] [CrossRef] [Green Version]
- Elnagdi, M.H.; Taha, N.H.; el All, F.A.M.A.; Abdel-Motaleb, R.M.; Mahmoud, F.F. Studies on condensed pyrazoles: Synthesis of new methyl and amino pyrazolo[1,5-a]pyrimidines and of pyrazolo[5,1-c][1,2,4]triazines. Collect. Czechoslov. Chem. Commun. 1989, 54, 1082–1091. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyrazole | R1 | R2 | % Tautomer (a) | % Tautomer (b) |
|---|---|---|---|---|
| 1 | Phenyl | Methyl | 66% | 34% |
| 2 | Phenyl | Ethyl | 63% | 37% |
| 3 | Phenyl | Trifluoromethyl | 27% | 73% |
| 4 | Phenyl | Isopropyl | 58% | 42% |
| 5 | Phenyl | Benzyl | 72% | 28% |
| 6 | Benzylphenyl | Trifluoromethyl | 0.6% | 99.4% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2020, 25, 42. https://doi.org/10.3390/molecules25010042
Secrieru A, O’Neill PM, Cristiano MLS. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules. 2020; 25(1):42. https://doi.org/10.3390/molecules25010042
Chicago/Turabian StyleSecrieru, Alina, Paul Michael O’Neill, and Maria Lurdes Santos Cristiano. 2020. "Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles" Molecules 25, no. 1: 42. https://doi.org/10.3390/molecules25010042
APA StyleSecrieru, A., O’Neill, P. M., & Cristiano, M. L. S. (2020). Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules, 25(1), 42. https://doi.org/10.3390/molecules25010042