
Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration †



Abstract
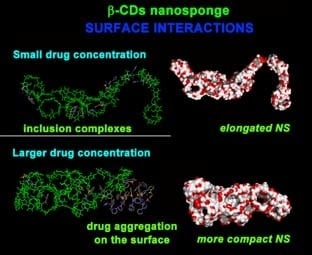
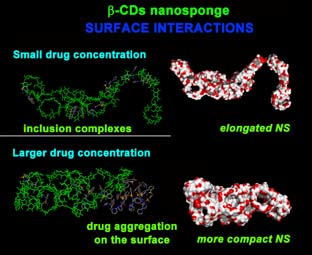
:
1. Introduction
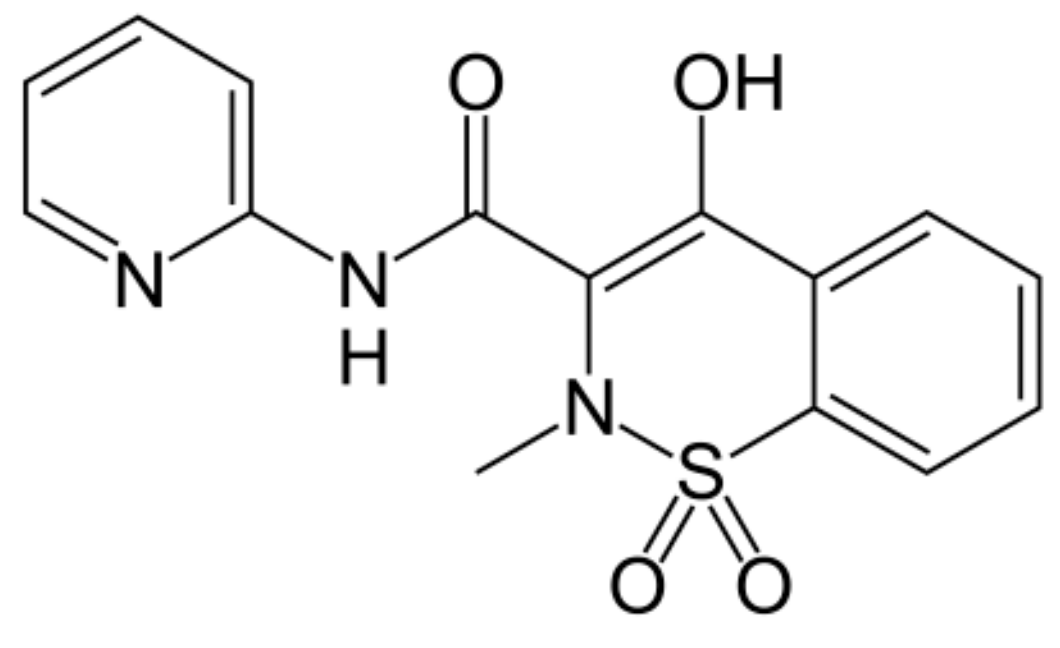
2. Results and Discussion
2.1. β-CD/PX Interaction
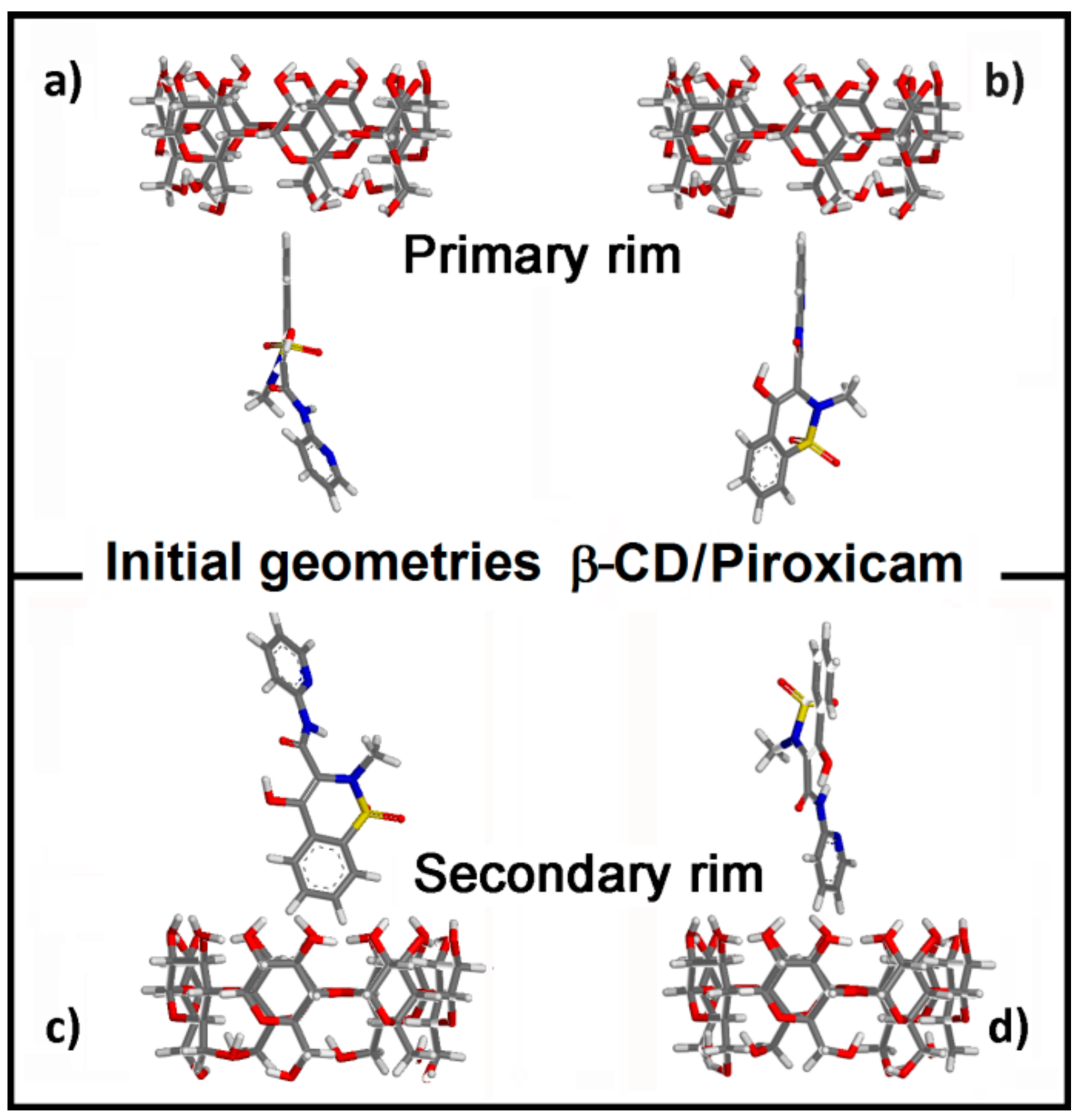
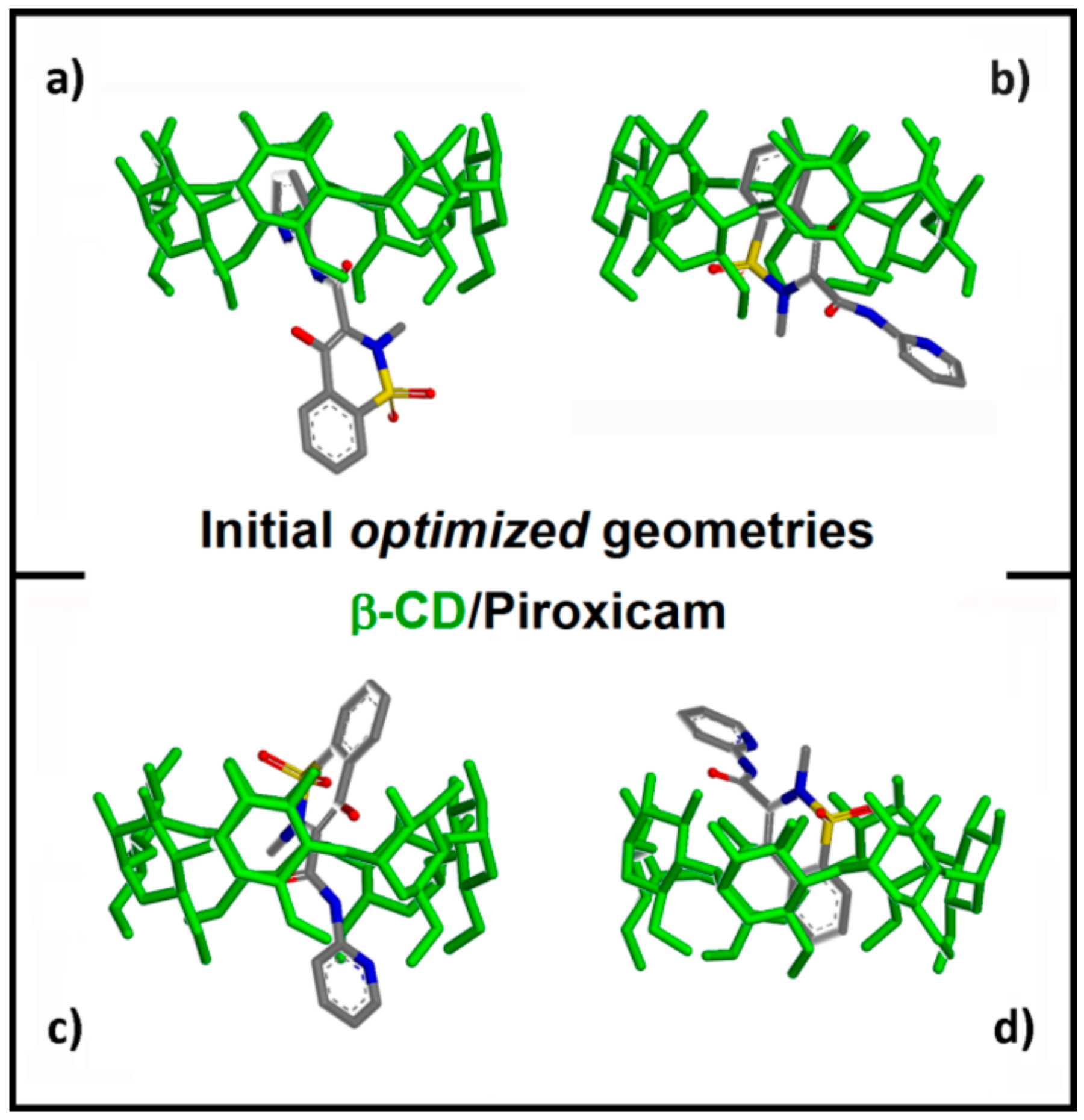
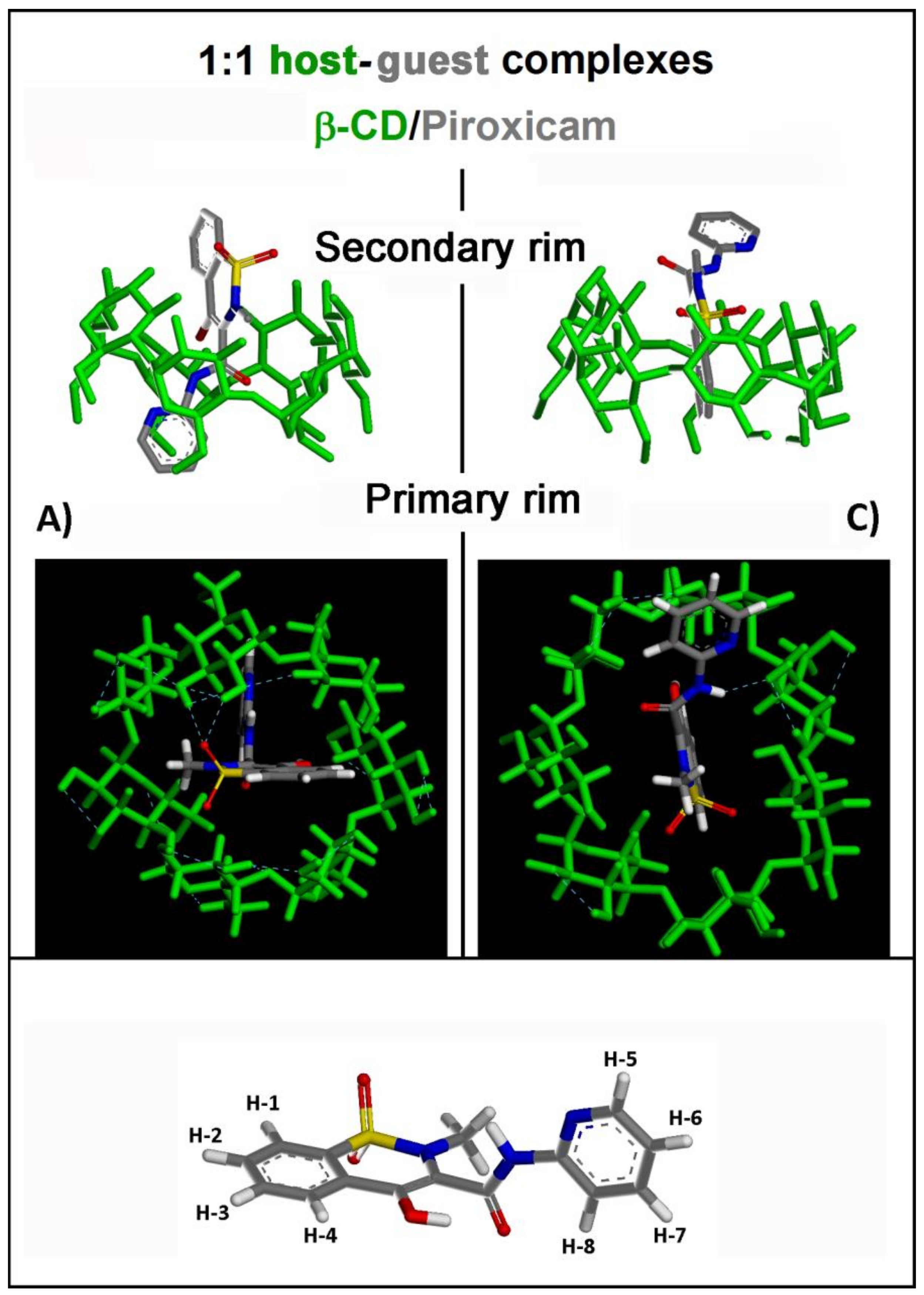
2.1.1. β-CD/PX Interaction: Complex Formation in a 1:1 Host–Guest Stoichiometry
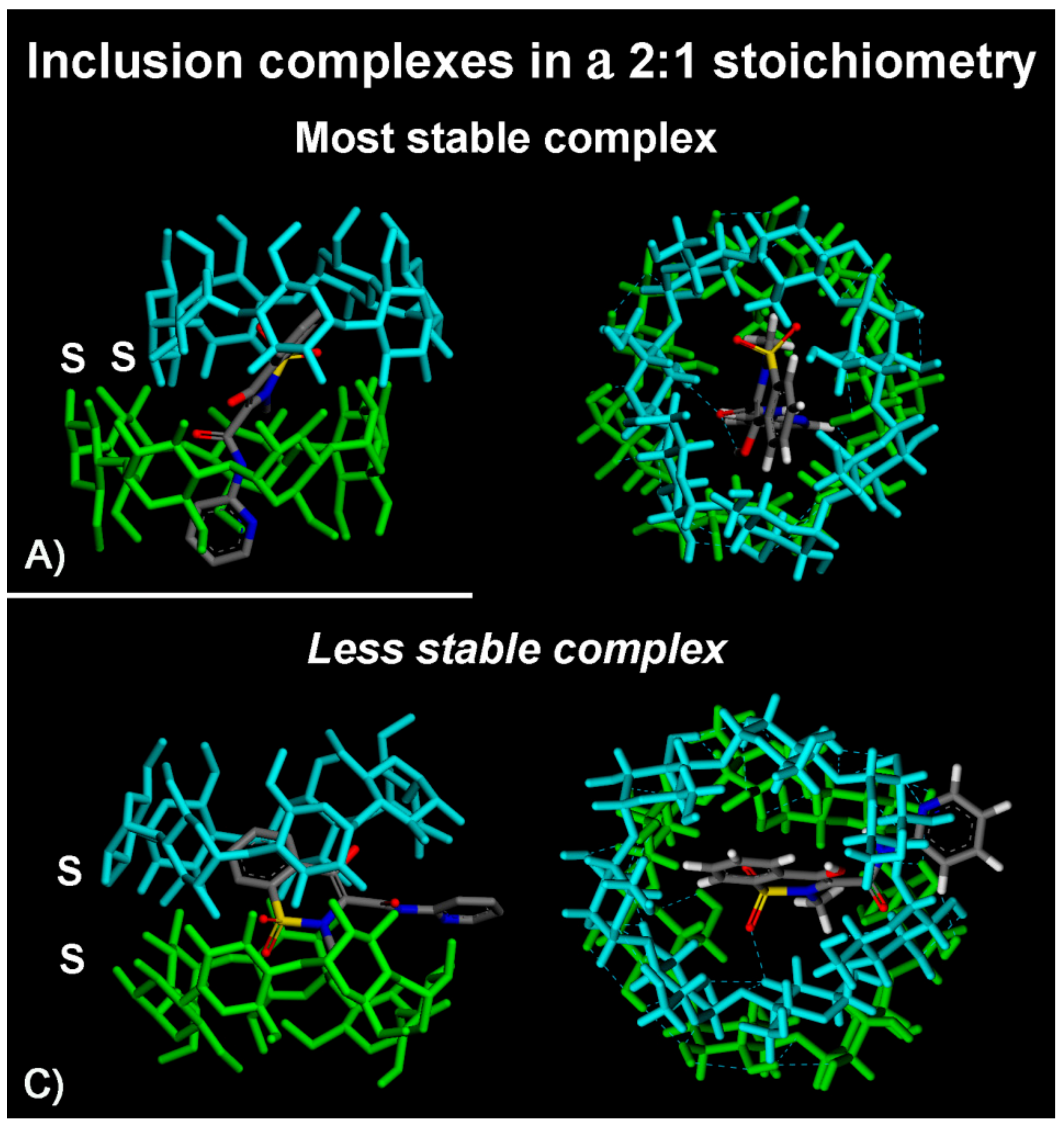
2.1.2. β-CD/PX Interaction: Complex Formation in a 2:1 Host–Guest Stoichiometry
2.2. PMA β-CD NS Model/PX Interaction at Different Concentrations
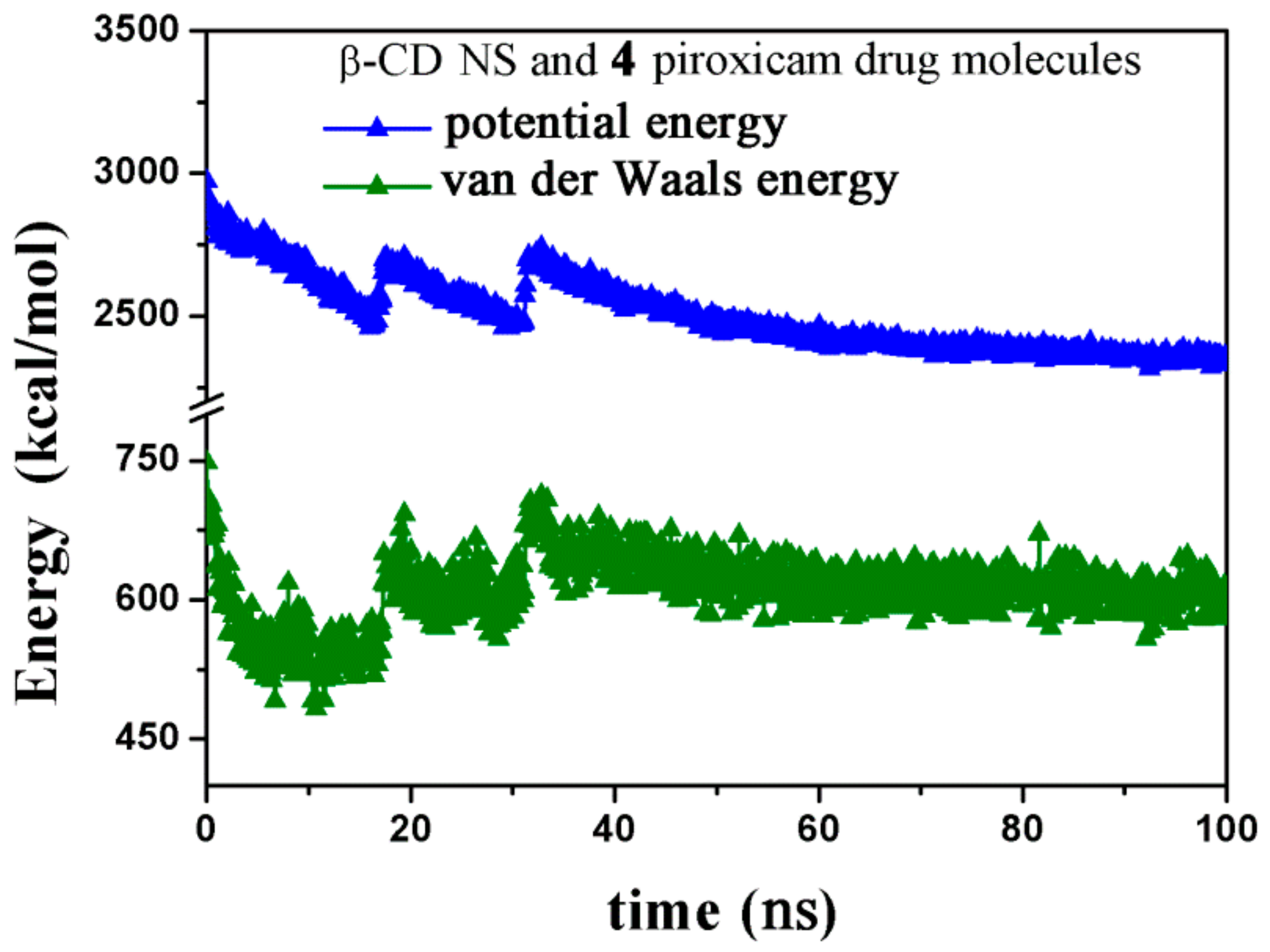
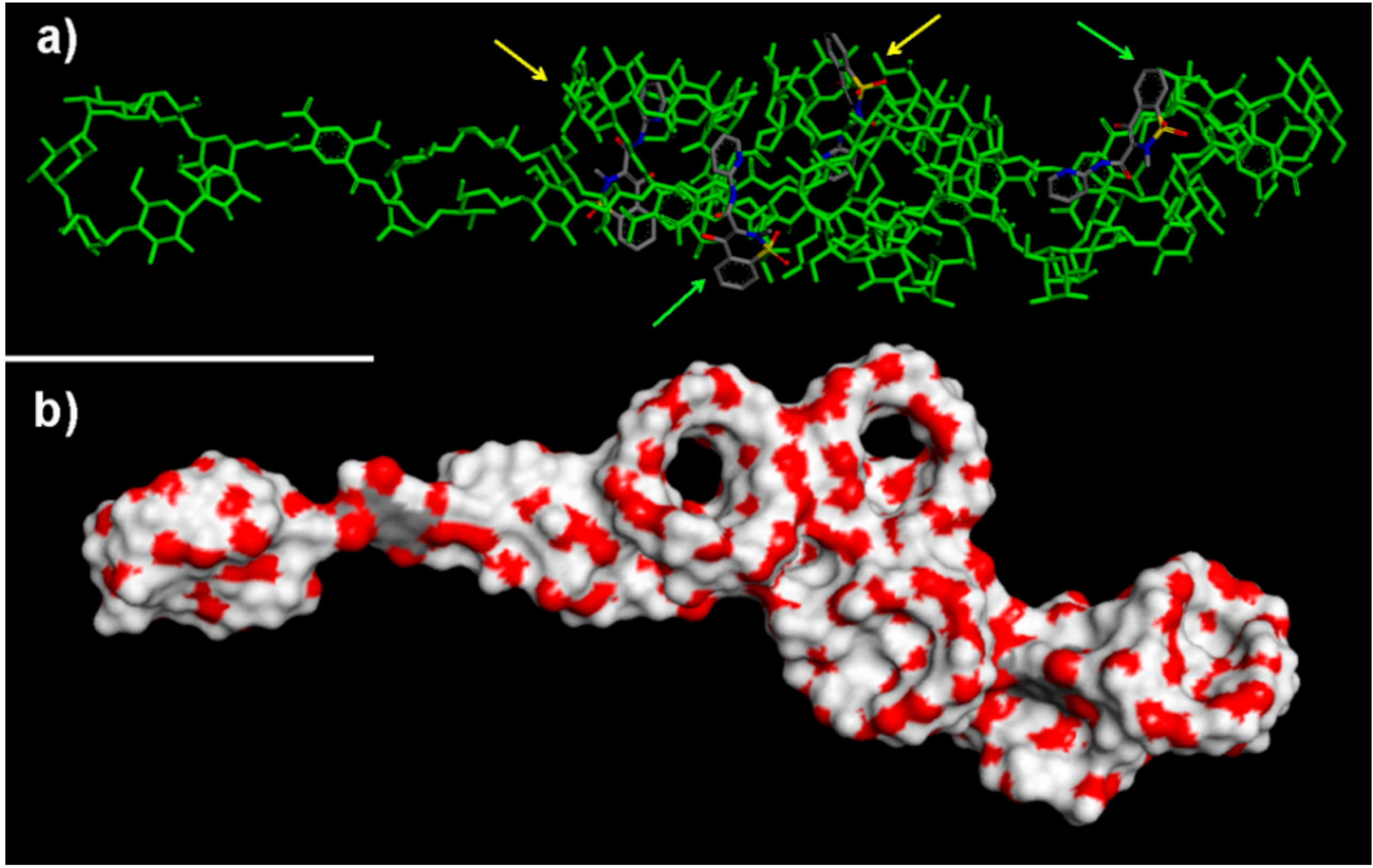
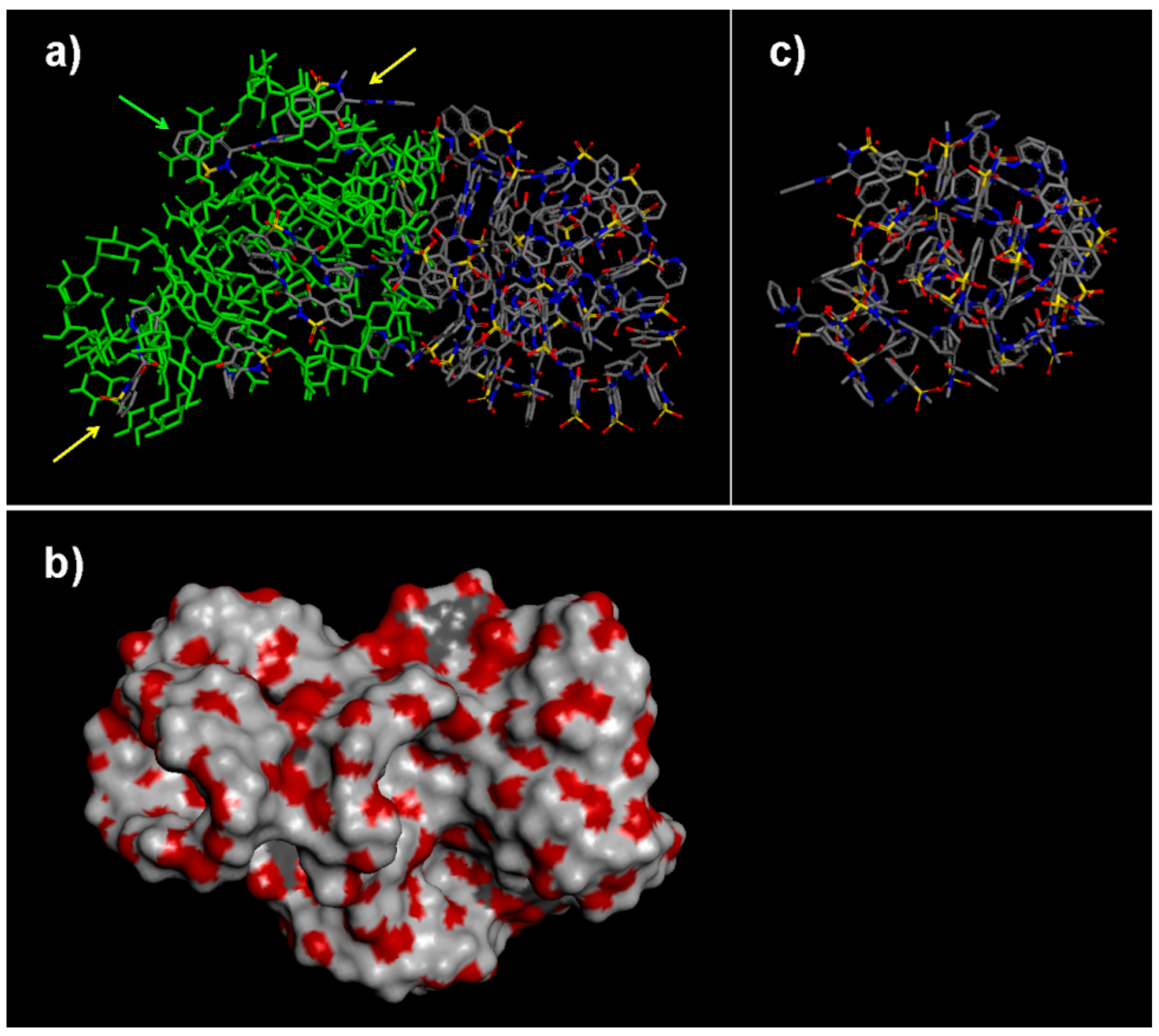
2.2.1. NS/PX Interaction: 8 β-CDs in the NS Model and 4 PX Molecules (β-CD/Drug in a 2:1 Stoichiometry)
2.2.2. NS/PX Interaction: 8 β-CDs in the NS Model and 8 PX Molecules (β-CD/Drug in a 1:1 Stoichiometry)
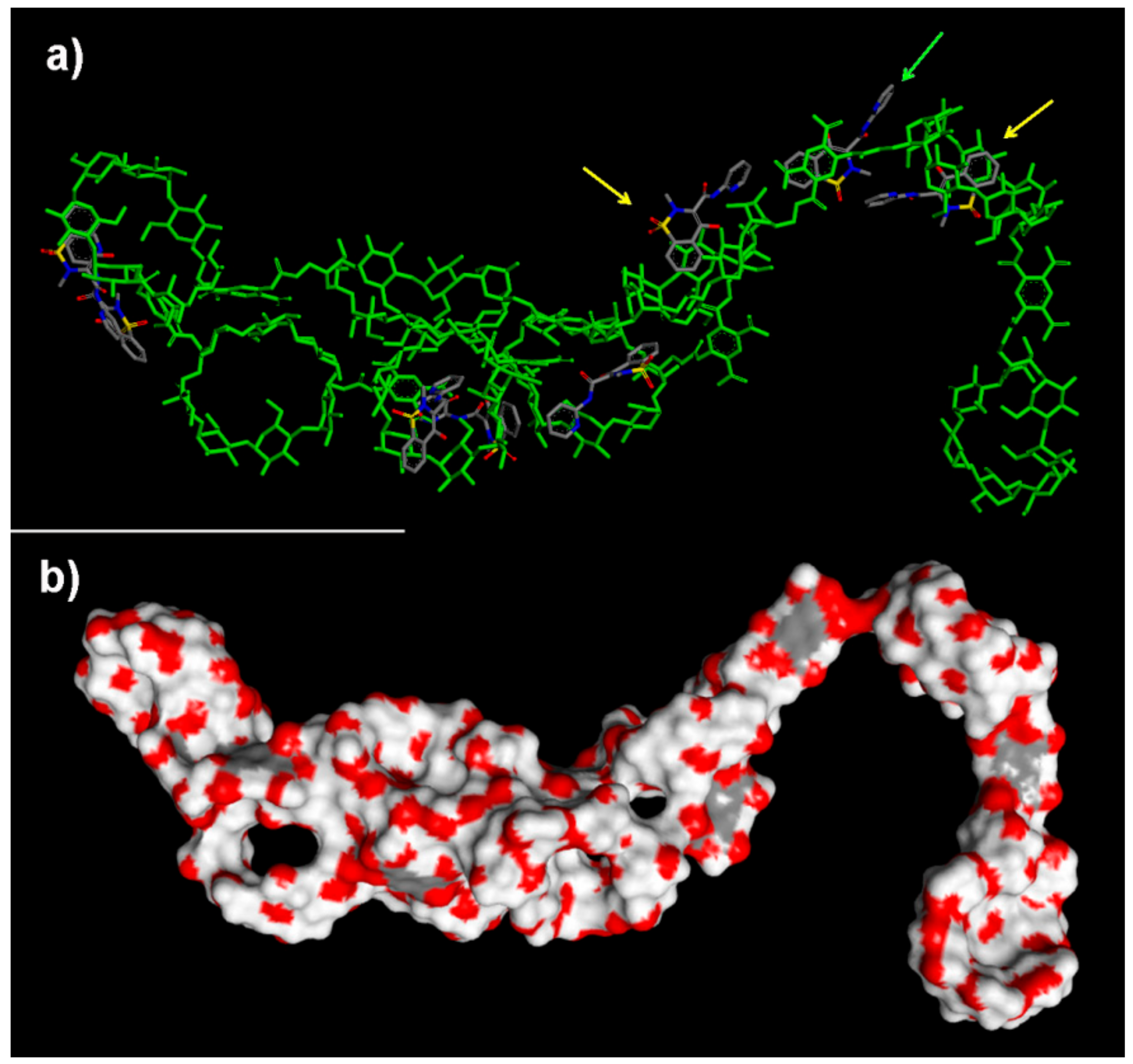
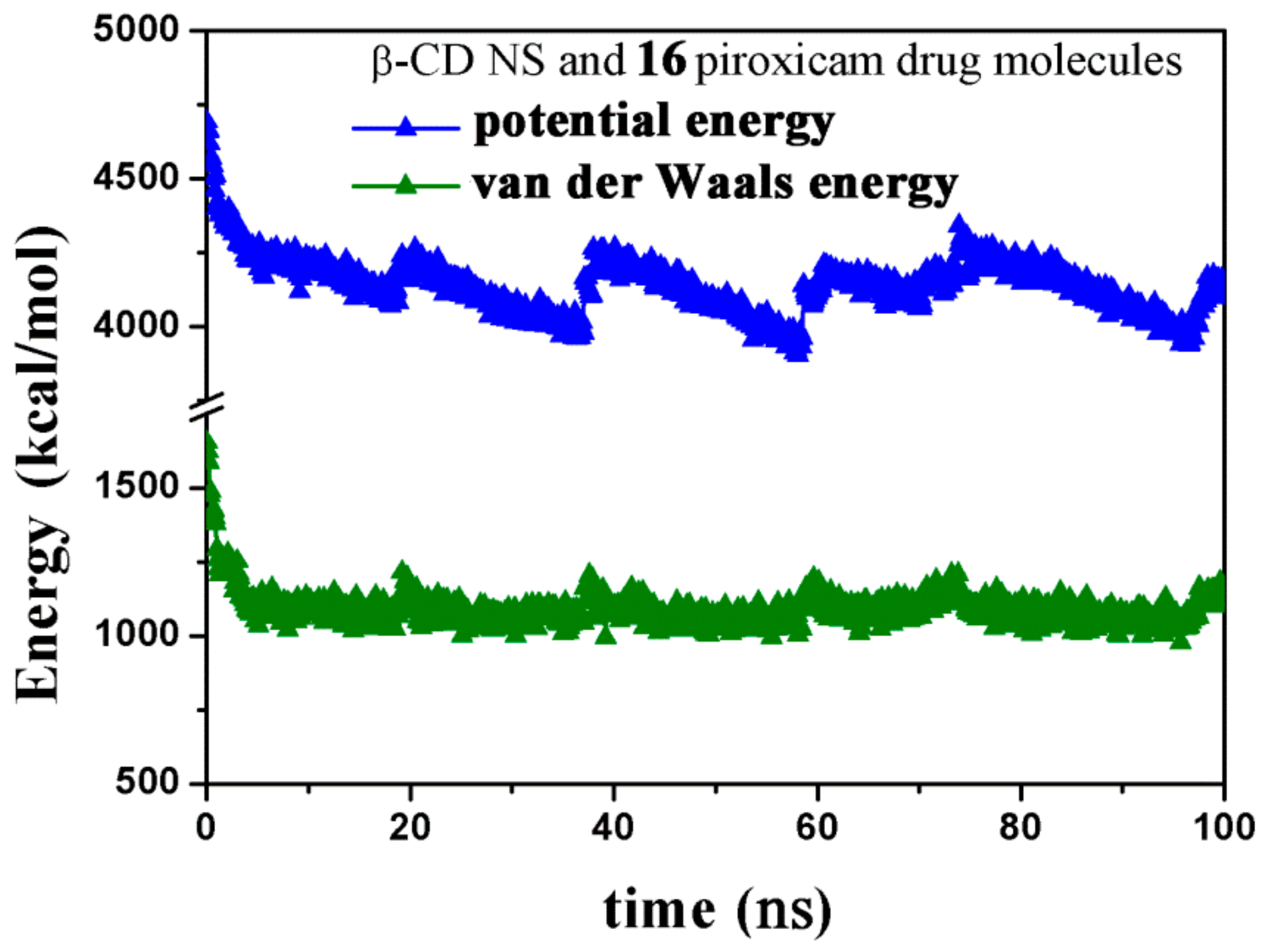
2.2.3. NS/PX Interaction: 8 β-CDs in the NS Model and 16 PX Molecules (β-CD/Drug in a 1:2 Stoichiometry)
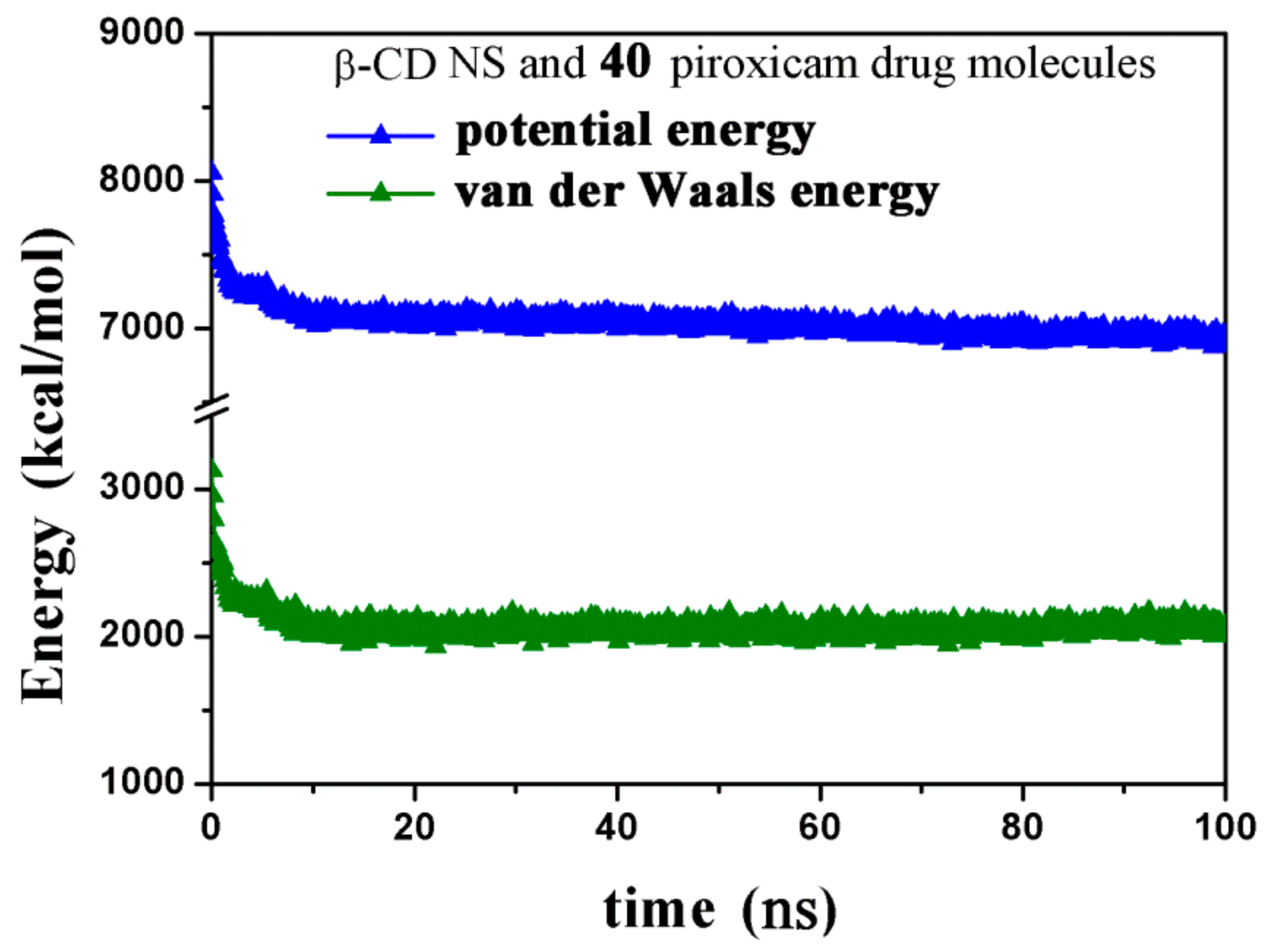
2.2.4. NS/PX Interaction: 8 β-CDs in the NS and 40 PX Molecules (β-CD/Drug in a 1:5 Stoichiometry)
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MM | molecular mechanics |
| MD | molecular dynamics |
| β-CD | β-cyclodextrin |
| PX | piroxicam |
| PMA | pyromellitic dianhydride |
| NS | nanosponge |
| β-CD NS | β-cyclodextrin nanosponge |
| NVT ensemble | Number of particles, Volume and Temperature are constant |
References
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Saraf, S.; Saraf, S. Cyclodextrin based novel drug delivery systems. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 23–42. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Stefansson, E. Cyclodextrins in ocular drug delivery: theoretical basis with dexamethasone as a sample drug. J. Drug Deliv. Sci. Technol. 2007, 17, 3–9. [Google Scholar] [CrossRef]
- Loftsson, T.; Stefánsson, E. Cyclodextrins and topical drug delivery to the anterior and posterior segments of the eye. Int. J. Pharm. 2017, 531, 413–423. [Google Scholar] [CrossRef]
- Szente, L.; Szejtli, J.; Kis, G.L. Spontaneous Opalescence of Aqueous γ-Cyclodextrin Solutions: Complex Formation or Self-Aggregation? J. Pharm. Sci. 1998, 87, 778–781. [Google Scholar] [CrossRef]
- Mele, A.; Mendichi, R.; Selva, A.; Molnar, P.; Toth, G. Non-covalent associations of cyclomaltooligosaccharides (cyclodextrins) with carotenoids in water. A study on the α- and β-cyclodextrin/ψ,ψ-carotene (lycopene) systems by light scattering, ionspray ionization and tandem mass spectrometry. Carbohydr. Res 2002, 337, 1129–1136. [Google Scholar] [CrossRef]
- Loftsson, T.; Saokham, P.; Couto, A.R.S. Self-association of cyclodextrins and cyclodextrin complexes in aqueous solutions. Int. J. Pharm. 2019, 560, 228–234. [Google Scholar] [CrossRef]
- Bondi’, M.L.; Scala, A.; Sortino, G.; Amore, E.; Botto, C.; Azzolina, A.; Balasus, D.; Cervello, M.; Mazzaglia, A. Nanoassemblies Based on Supramolecular Complexes of Nonionic Amphiphilic Cyclodextrin and Sorafenib as Effective Weapons to Kill Human HCC Cells. Biomacromolecules 2015, 16, 3784–3791. [Google Scholar] [CrossRef]
- Giglio, V.; Viale, M.; Bertone, V.; Maric, I.; Vaccarone, R.; Vecchio, G. Cyclodextrin polymers as nanocarriers for sorafenib. Investig. New Drugs 2017, 36, 370–379. [Google Scholar] [CrossRef]
- Swaminathan, S.; Vavia, P.; Trotta, F.; Torne, S. Formulation of betacyclodextrin based nanosponges of itraconazole. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 89–94. [Google Scholar] [CrossRef]
- Swaminathan, S.; Cavalli, R.; Trotta, F.; Ferruti, P.; Ranucci, E.; Gerges, I.; Manfredi, A.; Marinotto, D.; Vavia, P.R. In vitro release modulation and conformational stabilization of a model protein using swellable polyamidoamine nanosponges of β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2010, 68, 183–191. [Google Scholar] [CrossRef]
- Mele, A.; Castiglione, F.; Malpezzi, L.; Ganazzoli, F.; Raffaini, G.; Trotta, F.; Rossi, B.; Fontana, A.; Giunchi, G. HR MAS NMR, powder XRD and Raman spectroscopy study of inclusion phenomena in βCD nanosponges. J. Incl. Phenom. Macrocycl. Chem. 2010, 69, 403–409. [Google Scholar] [CrossRef]
- Trotta, F.; Mele, A. Nanosponges Synthesis and Applications; Wiley-VCH: Weinheim, Germany, 2019; ISBN 978-3-527-34099-6. [Google Scholar]
- Khuntawee, W.; Wolschann, P.; Rungrotmongkol, T.; Wong-Ekkabut, J.; Hannongbua, S. Molecular Dynamics Simulations of the Interaction of Beta Cyclodextrin with a Lipid Bilayer. J. Chem. Inf. Model. 2015, 55, 1894–1902. [Google Scholar] [CrossRef]
- Putaux, J.L.; Lancelon-Pi, C.; Legrand, F.-X.; Pastrello, M.; Choisnard, L.; Geze, A.; Rochas, C.; Wouessidjewe, D. Self-Assembly of Amphiphilic Biotranseterified beta-Cyclodextrins: Supramolecular Structure of Nanoparticles and Surface Properties. Langmuir 2017, 33, 7917–7928. [Google Scholar] [CrossRef]
- Fronza, G.; Mele, A.; Redenti, E.; Ventura, P. Proton Nuclear Magnetic Resonance Spectroscopy Studies of the Inclusion Complex of Piroxicam with β-Cyclodextrin. J. Pharm. Sci. 1992, 81, 1162–1165. [Google Scholar] [CrossRef]
- XiLiang, G.; Yu, Y.; Guoyan, Z.; Guomei, Z.; Jianbin, C.; ShaoMin, S. Study on inclusion interaction of piroxicam with beta-cyclodextrin derivatives. Spectrochim. Acta Part. A: Mol. Biomol. Spectrosc. 2003, 59, 14. [Google Scholar] [CrossRef]
- Dharmasthala, S.; Shabaraya, A.R.; Andrade, G.S.; Shriram, R.G.; Hebbar, S.; Dubey, A. Fast Dissolving Oral Film of Piroxicam: Solubility Enhancement by forming an Inclusion Complex with β-cyclodextrin, Formulation and Evaluation. J. Young- Pharm. 2018, 11, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Raffaini, G.; Ganazzoli, F.; Malpezzi, L.; Fuganti, C.; Fronza, G.; Panzeri, W.; Mele, A. Validating a Strategy for Molecular Dynamics Simulations of Cyclodextrin Inclusion Complexes through Single-Crystal X-ray and NMR Experimental Data: A Case Study. J. Phys. Chem. B 2009, 113, 9110–9122. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Molecular dynamics study of host–guest interactions in cyclodextrins: methodology and data analysis for a comparison with solution data and the solid-state structure. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 683–688. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Hydration and flexibility of α-, β-, γ- and δ-cyclodextrin: a molecular dynamics study. Chem. Phys. 2003, 333, 625–635. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Surface hydration of polymeric (bio)materials: A molecular dynamics simulation study. J. Biomed. Mater. Res. Part. A 2009, 9999, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Raffaini, G.; Ganazzoli, F. Protein adsorption on biomaterial and nanomaterial surfaces: a molecular modeling approach to study non-covalent interactions. J. Appl. Biomater. Biomech. 2011, 8, 135–145. [Google Scholar] [CrossRef]
- Raffaini, G.; Mazzaglia, A.; Ganazzoli, F. Aggregation behaviour of amphiphilic cyclodextrins: the nucleation stage by atomistic molecular dynamics simulations. Beilstein J. Org. Chem. 2015, 11, 2459–2473. [Google Scholar] [CrossRef] [Green Version]
- Raffaini, G.; Ganazzoli, F.; Mazzaglia, A. Aggregation behavior of amphiphilic cyclodextrins in a nonpolar solvent: evidence of large-scale structures by atomistic molecular dynamics simulations and solution studies. Beilstein J. Org. Chem. 2016, 12, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Raffaini, G.; Ganazzoli, F. A Molecular Dynamics Study of the Inclusion Complexes of C60with Some Cyclodextrins. J. Phys. Chem. B 2010, 114, 7133–7139. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. A Molecular Dynamics Study of a Photodynamic Sensitizer for Cancer Cells: Inclusion Complexes of γ-Cyclodextrins with C70. Int. J. Mol. Sci. 2019, 20, 4831. [Google Scholar] [CrossRef] [Green Version]
- Mele, A.; Ganazzoli, F.; Raffaini, G.; Juza, M.; Schurig, V. Macrocycle conformation and self-inclusion phenomena in octakis(3-O-butanoyl-2,6-di-O-n-pentyl)-γ-cyclodextrin (Lipodex E) by NMR spectroscopy and molecular dynamics. Carbohydr. Res. 2003, 338, 625–635. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F.; Mele, A.; Castiglione, F. A molecular dynamics study of cyclodextrin nanosponge models. J. Incl. Phenom. Macrocycl. Chem. 2012, 75, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Pean, C.; Djedaïni-Pilard, F.; Perly, B. Reliable NMR Experiments for the Determination of the Structure of Cyclodextrin Inclusion Complexes in Solution. In Proceedings of the Ninth International Symposium on Cyclodextrins; Labandeira, J.J.T., Vila-Jato, J.L., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 659–662. [Google Scholar]
- Landy, D. Measuring Binding Constants of Cyclodextrin Inclusion Compounds. In Advanced Nanostructured Materials for Environmental Remediation; Fourmentin, S., Crini, G., Lichtfouse, E., Eds.; Cyclodextrin Fundamentals, Reactivity and Analysis; Springer International Publishing: Cham, Switzerland, 2018; pp. 223–255, (Environmental Chemistry for a Sustainable World); Available online: https://doi.org/10.1007/978-3-319-76159-6_5.
- Hwang, M.-J.; Ni, X.; Waldman, M.; Ewig, C.S.; Hagler, A.T. Derivation of class II force fields. VI. Carbohydrate compounds and anomeric effects. Biopolym. 1998, 45, 435–468. [Google Scholar] [CrossRef]
- Materials Studio BIOVIA. Accelrys Inc. InsightII 2000; Accelrys Inc.: San Diego, CA, USA, 2000. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geometry | Epot (kJ/mol) | Eint (kJ/mol) | Intermolecular H-Bonds | Intramolecular β-CD H-Bonds |
|---|---|---|---|---|
| A) Most stable | 766.5 | −173.2 | 1 bidentate (with II rim) | 16 |
| B) Metastable | 769.4 | −170.3 | 1 (with I rim) | 17 |
| C) Less stable | 776.6 | −163.1 | 1 (with II rim) | 8 |
| D) Metastable | 771.5 | −168.1 | 1 bidentate (with II rim) | 12 |
| Geometry | Epot (kJ/mol) | Eint (kJ/mol) | Intermolecular β-CD/PX H-bonds | Intermolecular β-CD H-bonds | Intramol.first β-CD H-bonds | Intramol.second β-CD H-bonds |
|---|---|---|---|---|---|---|
| A) Most stable | 233.4 | −381.7 | 1 (with II rim) | 5 | 9 | 8 |
| B) Less stable | 247.6 | −322.8 | 2 (with II rim) | 4 | 18 | 15 |
| C) Metastable | 241.4 | −348.8 | 1 (with II rim) | 3 | 13 | 15 |
| D) Metastable | 242.8 | −342.5 | 2 (1 with I and II rims) | 5 | 16 | 18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffaini, G.; Ganazzoli, F. Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration. Molecules 2020, 25, 2848. https://doi.org/10.3390/molecules25122848
Raffaini G, Ganazzoli F. Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration. Molecules. 2020; 25(12):2848. https://doi.org/10.3390/molecules25122848
Chicago/Turabian StyleRaffaini, Giuseppina, and Fabio Ganazzoli. 2020. "Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration" Molecules 25, no. 12: 2848. https://doi.org/10.3390/molecules25122848
APA StyleRaffaini, G., & Ganazzoli, F. (2020). Understanding Surface Interaction and Inclusion Complexes between Piroxicam and Native or Crosslinked β-Cyclodextrins: The Role of Drug Concentration. Molecules, 25(12), 2848. https://doi.org/10.3390/molecules25122848