
The Role of State-of-the-Art Quantum-Chemical Calculations in Astrochemistry: Formation Route and Spectroscopy of Ethanimine as a Paradigmatic Case

 , , , and
, , , and 
Abstract
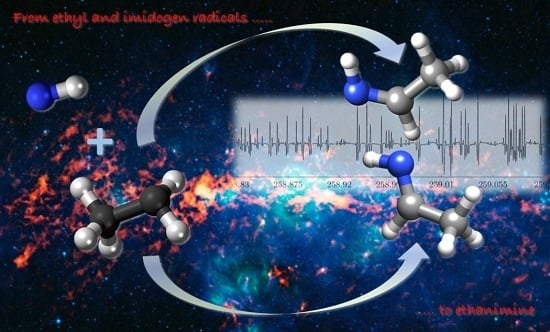
:
1. Introduction
- (1)
- The investigation of reactive potential energy surfaces (PESs) from both energetic (thermochemistry) and kinetic points of view. Two possibilities can be actually envisaged: (i) starting from purposely chosen precursors the formation route of the sought product (i.e., a molecule already identified in the ISM) is derived; (ii) starting from small reactive species the possible pathways are elaborated. In both cases, the harsh conditions of the ISM (extremely cold—down to 10 K—regions, extremely low density—even 10 particles/cm) are used as constraints.
- (2)
- Referring to point (1.ii), the accurate prediction of the spectroscopic parameters of those products that can be of interest and for which such information is still missing is carried out.The second piece of information (point (2)) is then used to guide and support laboratory measurements that, in the field of rotational spectroscopy, are mandatory to further proceed toward astronomical searches. Finally, astronomical data can confirm the plausibility of the developed formation pathway. For example, the identification (in astronomical surveys) of the sought product of the reaction investigated can provide an indirect confirmation. Astronomical observations can also be used to support the effectiveness of a gas-phase formation route [25]. Furthermore, as in the case of ethanimine, that is, in the presence of two different isomers, the computed branching ratios can be compared with the relative astronomical abundance.
1.1. The Thermochemistry-Kinetics-Spectroscopy Strategy
1.1.1. Formation Pathway
1.1.2. Spectroscopic Characterization
1.2. Organization of the Paper
2. Computational Methodology
2.1. Electronic Structure Calculations
2.1.1. Reactive Potential Energy Surface
2.1.2. Spectroscopic Parameters
2.1.3. Composite Schemes
The CC-Based Approaches
The ChS Approach
2.2. Kinetic Models
3. Results and Discussion
3.1. The NH + C2H5 Reaction: Thermochemistry
3.2. The NH + C2H5 Reaction: Rate Coefficients
3.3. E-/Z-CH3CHNH: Spectroscopic Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snyder, L.E.; Buhl, D.; Zuckerman, B.; Palmer, P. Microwave detection of interstellar formaldehyde. Phys. Rev. Lett. 1969, 22, 679. [Google Scholar] [CrossRef]
- Ball, J.A.; Gottlieb, C.A.; Lilley, A.E.; Radford, H.E. Detection of Methyl Alcohol in Sagittarius. Astrophys. J. Lett. 1970, 162, L203. [Google Scholar] [CrossRef]
- Zuckerman, B.; Ball, J.A.; Gottlieb, C.A. Microwave detection of interstellar formic acid. Astrophys. J. 1971, 163, L41. [Google Scholar] [CrossRef]
- McGuire, B.A. Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. Suppl. Ser. 2018, 239, 17. [Google Scholar] [CrossRef]
- Tielens, A.G.G.M. The molecular universe. Rev. Mod. Phys. 2013, 85, 1021–1081. [Google Scholar] [CrossRef]
- Herbst, E.; Dishoeck, E. Complex Interstellar Organic Molecules. Annu. Rev. Astron. Astrophys. 2009, 47, 427. [Google Scholar] [CrossRef]
- Hollis, J.M.; Vogel, S.N.; Snyder, L.E.; Jewell, P.R.; Lovas, F.J. Interstellar glycolaldehyde: The first sugar. Astrophys. J. 2000, 540, L107. [Google Scholar] [CrossRef]
- Hollis, J.M.; Lovas, F.J.; Remijan, A.R.; Jewell, P.R.; Ilyushin, V.V.; Kleiner, I. Detection of Acetamide (CH3CONH2): The Largest Interstellar Molecule with a Peptide Bond. Astrophys. J. 2006, 643, L25. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, J.K.; Favre, C.; Bisschop, S.E.; Bourke, T.L.; van Dischoek, E.F.; Schmalzl, M. Detection of the Simplest Sugar, Glycolaldehyde, in a Solar-type Protostar with ALMA. Astrophys. J. 2012, 757, L4. [Google Scholar] [CrossRef]
- Zaleski, D.P.; Seifert, N.A.; Steber, A.L.; Muckle, M.T.; Loomis, R.A.; Corby, J.F.; Martinez, O.J.; Carbtree, K.N.; Jewell, F.R.; Hollis, J.M.; et al. Detection of E-Cyanomethanimine Toward Sagittarius B2(N) in the Green Bank Telescope PRIMOS Survey. Astrophys. J. 2013, 765, L10. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Puzzarini, C. Quantum Chemistry Meets Spectroscopy for Astrochemistry: Increasing Complexity toward Prebiotic Molecules. ACC Chem. Res. 2015, 48, 1413. [Google Scholar] [CrossRef] [PubMed]
- Garrod, R.T.; Belloche, A.; Müller, H.S.P.; Menten, K.M. Exploring molecular complexity with ALMA (EMoCA): Simulations of branched carbon-chain chemistry in Sgr B2(N). Astron. Astrophys. 2017, 601, A48. [Google Scholar] [CrossRef] [Green Version]
- Melosso, M.; Melli, A.; Puzzarini, C.; Codella, C.; Spada, L.; Dore, L.; Esposti, C.D.; Lefloch, B.; Bachiller, R.; Ceccarelli, C.; et al. Laboratory measurements and astronomical search for cyanomethanimine. Astron. Astrophys. 2018, 609, A121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belloche, A.; Garrod, R.T.; Müller, H.S.P.; Menten, K.M.; Medvedev, I.; Thomas, J.; Kisiel, Z. Re-exploring Molecular Complexity with ALMA (ReMoCA): Interstellar detection of urea. Astron. Astrophys. 2019, 628, A10. [Google Scholar] [CrossRef] [Green Version]
- Garrod, R.T.; Herbst, E. Formation of methyl formate and other organic species in the warm-up phase of hot molecular cores. Astron. Astrophys. 2006, 457, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Garrod, R.T.; Weaver, S.L.W.; Herbst, E. Complex Chemistry in Star-forming Regions: An Expanded Gas-Grain Warm-up Chemical Model. Astrophys. J. 2008, 682, 283–302. [Google Scholar] [CrossRef] [Green Version]
- Balucani, N.; Ceccarelli, C.; Taquet, V. Formation of complex organic molecules in cold objects: The role of gas-phase reactions. Mon. Not. R. Astron. Soc. 2015, 449, L16–L20. [Google Scholar] [CrossRef] [Green Version]
- Linnartz, H.; Ioppolo, S.; Fedoseev, G. Atom addition reactions in interstellar ice analogues. Int. Rev. Phys. Chem. 2015, 34, 205–237. [Google Scholar] [CrossRef] [Green Version]
- Vazart, F.; Calderini, D.; Puzzarini, C.; Skouteris, D.; Barone, V. State-of-the-Art Thermochemical and Kinetic Computations for Astrochemical Complex Organic Molecules: Formamide Formation in Cold Interstellar Clouds as a Case Study. J. Chem. Theory Comput. 2016, 12, 5385–5397. [Google Scholar] [CrossRef]
- Rimola, A.; Skouteris, D.; Balucani, N.; Ceccarelli, C.; Enrique-Romero, J.; Taquet, V.; Ugliengo, P. Can Formamide Be Formed on Interstellar Ice? An Atomistic Perspective. ACS Earth Space Chem. 2018, 2, 720–734. [Google Scholar] [CrossRef]
- Vastel, C.; Ceccarelli, C.; Lefloch, B.; Bachiller, R. The origin of complex organic molecules in prestellar cores. Astrophys. J. 2014, 795, L2. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S. (Ed.) Introduction to Astrochemistry (Chemical Evolution from Interstellar Clouds to Star and Planet Formation); Springer Japan KK: Tokyo, Japan, 2017. [Google Scholar]
- Puzzarini, C. Grand Challenges in Astrochemistry. Front. Astron. Space Sci. 2020, 7, 19. [Google Scholar] [CrossRef]
- Puzzarini, C.; Barone, V. The challenging playground of astrochemistry: An integrated rotational spectroscopy–quantum chemistry strategy. Phys. Chem. Chem. Phys. 2020, 22, 6507–6523. [Google Scholar] [CrossRef] [PubMed]
- Codella, C.; Ceccarelli, C.; Caselli, P.; Balucani, N.; Barone, V.; Fontani, F.; Lefloch, B.; Podio, L.; Viti, S.; Feng, S.; et al. Seeds of Life in Space (SOLIS): II. Formamide in protostellar shocks: Evidence for gas-phase formation. Astron. Astrophys. 2017, 605, L3. [Google Scholar] [CrossRef]
- Quan, D.; Herbst, E.; Corby, J.F.; Durr, A.; Hassel, G. Chemical simulations of prebiotic molecules: Interstellar ethanimine isomers. Astrophys. J. 2016, 824, 129. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E. Pathways to Glycine and Other Amino Acids in Ultraviolet-irradiated Astrophysical Ices Determined via Quantum Chemical Modeling. Astrophys. J. 2002, 571, L177–L180. [Google Scholar] [CrossRef] [Green Version]
- Elsila, J.E.; Dworkin, J.P.; Bernstein, M.P.; Martin, M.P.; Sandford, S.A. Mechanisms of amino acid formation in interstellar ice analogs. Astrophys. J. 2007, 660, 911. [Google Scholar] [CrossRef] [Green Version]
- Loomis, R.A.; Zaleski, D.P.; Steber, A.L.; Neill, J.L.; Muckle, M.T.; Harris, B.J.; Hollis, J.M.; Jewell, P.R.; Lattanzi, V.; Lovas, F.J.; et al. The detection of interstellar ethanimine (CH3CHNH) from observations taken during the GBT PRIMOS survey. Astrophys. J. Lett. 2013, 765, L9. [Google Scholar] [CrossRef] [Green Version]
- Balucani, N.; Leonori, F.; Petrucci, R.; Stazi, M.; Skouteris, D.; Rosi, M.; Casavecchia, P. Formation of nitriles and imines in the atmosphere of Titan: Combined crossed-beam and theoretical studies on the reaction dynamics of excited nitrogen atoms N(2D) with ethane. Faraday Discuss. 2010, 147, 189–216. [Google Scholar] [CrossRef]
- Clarke, D.; Ferris, J.P. Chemical evolution on Titan: Comparisons to the prebiotic Earth. Orig. Life Evol. Biol. 1997, 27, 225–248. [Google Scholar] [CrossRef]
- Raulin, F.; McKay, C.; Lunine, J.; Owen, T. (Eds.) Titan from Cassini-Huygens; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Salta, Z.; Tasinato, N.; Lupi, J.; Boussessi, R.; Balbi, A.; Puzzarini, C.; Barone, V. Exploring the Maze of C2N2H5 Radicals and Their Fragments in the Interstellar Medium with the Help of Quantum-Chemical Computations. ACS Earth Space Chem. 2020, 4, 774–782. [Google Scholar] [CrossRef]
- Barone, V.; Latouche, C.; Skouteris, D.; Vazart, F.; Balucani, N.; Ceccarelli, C.; Lefloch, B. Gas-phase formation of the prebiotic molecule formamide: Insights from new quantum computations. Mon. Not. R. Astron. Soc. Lett. 2015, 453, L31–L35. [Google Scholar] [CrossRef] [Green Version]
- Skouteris, D.; Balucani, N.; Ceccarelli, C.; Vazart, F.; Puzzarini, C.; Barone, V.; Codella, C.; Lefloch, B. The Genealogical Tree of Ethanol: Gas-phase Formation of Glycolaldehyde, Acetic Acid, and Formic Acid. Astrophys. J. 2018, 854, 135. [Google Scholar] [CrossRef]
- Puzzarini, C.; Bloino, J.; Tasinato, N.; Barone, V. Accuracy and Interpretability: The Devil and the Holy Grail. New Routes across Old Boundaries in Computational Spectroscopy. Chem. Rev. 2019, 119, 8131–8191. [Google Scholar] [CrossRef] [PubMed]
- Skouteris, D.; Vazart, F.; Ceccarelli, C.; Balucani, N.; Puzzarini, C.; Barone, V. New quantum chemical computations of formamide deuteration support gas-phase formation of this prebiotic molecule. Mon. Not. R. Astron. Soc. Lett. 2017, 468, L1–L5. [Google Scholar] [CrossRef]
- Suzuki, T.; Ohishi, M.; Hirota, T.; Saito, M.; Majumdar, L.; Wakelam, V. Survey observations of a possible glycine precursor, methanimine (CH2NH). Astrophys. J. 2016, 825, 79. [Google Scholar] [CrossRef] [Green Version]
- Balucani, N.; Skouteris, D.; Ceccarelli, C.; Codella, C.; Falcinelli, S.; Rosi, M. A theoretical investigation of the reaction between the amidogen, NH, and the ethyl, C2H5, radicals: A possible gas-phase formation route of interstellar and planetary ethanimine. Mol. Astrophys. 2018, 13, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K.; Tandan, P.; Misra, A. A quantum chemical study on the formation of ethanimine (CH3CHNH) in the interstellar ice. Astrophys. Space Sci. 2018, 363, 213. [Google Scholar] [CrossRef]
- Brown, D.E.; Godfry, P.D.; Winkler, D.A. The microwave spectrum of (Z)-Ethanimine. Aust. J. Chem. 1980, 33, 1. [Google Scholar] [CrossRef]
- Lovas, F.J.; Suenram, R.D.; Johnson, D.R.; Clark, F.O.; Tiemann, F. Pyrolysis of ethylamine. II. Synthesis and microwave spectrum of ethylidenimine (CH3CH=NH). J. Chem. Phys. 1980, 72, 4964–4972. [Google Scholar] [CrossRef]
- Melli, A.; Melosso, M.; Tasinato, N.; Bosi, G.; Spada, L.; Bloino, J.; Mendolicchio, M.; Dore, L.; Barone, V.; Puzzarini, C. Rotational and Infrared Spectroscopy of Ethanimine: A Route toward Its Astrophysical and Planetary Detection. Astrophys. J. 2018, 855, 13. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, K.; Hamada, Y.; Tsuboi, M.; Koga, Y.; Kondo, S. Pyrolysis of amines: Infrared spectrum of ethylideneimine. J. Mol. Spectr. 1984, 105, 81–92. [Google Scholar] [CrossRef]
- Stolkin, I.; Ha, T.K.; Günthard, H. N-methylmethyleneimine and ethylideneimine: Gas- and matrix-infrared spectra, Ab initio calculations and thermodynamic properties. Chem. Phys. 1977, 21, 327–347. [Google Scholar] [CrossRef]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 014108. [Google Scholar] [CrossRef]
- Klippenstein, S.J. From Theoretical Reaction Dynamics to Chemical Modeling of Combustion. Proc. Combust. Inst. 2017, 36, 77–111. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package v2.1. Available online: http://www.cfour.de (accessed on 17 January 2020).
- Kállay, M.; Nagy, P.R.; Rolik, Z.; Mester, D.; Samu, G.; Csontos, J.; Csóka, J.; Szabó, B.P.; Gyevi-Nagy, L.; Ladjánszki, I.; et al. MRCC, a Quantum Chemical Program Suite. 2018. Available online: http://www.mrcc.hu (accessed on 17 January 2020).
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zheng, Y.; Melli, A.; Spada, L.; Lu, T.; Feng, G.; Gou, Q.; Barone, V.; Puzzarini, C. Theory meets experiment for elucidating the structure and stability of non-covalent complexes: Water–amine interaction as a proof of concept. Phys. Chem. Chem. Phys. 2020, 22, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Spada, L.; Chen, J.; Gao, S.; Alessandrini, S.; Feng, G.; Puzzarini, C.; Gou, Q.; Grabow, J.U.; Barone, V. The Unexplored World of Cycloalkene–Water Complexes: Primary and Assisting Interactions Unraveled by Experimental and Computational Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 13935–13941. [Google Scholar] [CrossRef] [PubMed]
- Boussessi, R.; Geselin, G.; Tasinato, N.; Barone, V. DFT meets the segmented polarization consistent basis sets: Performances in the computation of molecular structures, rotational and vibrational spectroscopic properties. J. Mol. Struct. 2020, 1208, 127886. [Google Scholar] [CrossRef]
- Spada, L.; Tasinato, N.; Vazart, F.; Barone, V.; Caminati, W.; Puzzarini, C. Noncovalent Interactions and Internal Dynamics in Pyridine–Ammonia: A Combined Quantum-Chemical and Microwave Spectroscopy Study. Chem. Eur. J. 2017, 23, 4876–4883. [Google Scholar] [CrossRef] [PubMed]
- Spada, L.; Tasinato, N.; Bosi, G.; Vazart, F.; Barone, V.; Puzzarini, C. On the competition between weak O−H···F and C−H···F hydrogen bonds, in cooperation with C−H···O contacts, in the difluoromethane–tert-butyl alcohol cluster. J. Mol. Spectrosc. 2017, 337, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupi, J.; Puzzarini, C.; Cavallotti, C.; Barone, V. State-of-the-art quantum chemistry meets variable reaction coordinate transition state theory to solve the puzzling case of the H2S + Cl system. J. Chem. Theory Comput. 2020. submitted. [Google Scholar]
- Puzzarini, C.; Salta, Z.; Tasinato, N.; Lupi, J.; Cavallotti, C.; Barone, V. A twist on the reaction of the CN radical with methylamine in the interstellar medium: New hints from a state-of-the-art quantum-chemical study. Mon. Not. R. Astron. Soc. Lett. 2020. submitted. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Rajvanshi, J.S.; Baluja, K.L. Electron-impact study of the NH radical using the R-matrix method. Phys. Rev. A 2010, 82, 062710. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Lee, T.J.; Taylor, P.R.; François, J. The anharmonic force field of ethylene, C2H4, by means of accurate ab initio calculations. J. Chem. Phys. 1995, 103, 2589–2602. [Google Scholar] [CrossRef] [Green Version]
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. J. Chem. Phys. 2004, 120, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Bloino, J.; Biczysko, M.; Barone, V. General Perturbative Approach for Spectroscopy, Thermodynamics, and Kinetics: Methodological Background and Benchmark Studies. J. Chem. Theor. Comput. 2012, 8, 1015–1036. [Google Scholar] [CrossRef] [PubMed]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra, 3rd ed.; John Wiley & Sons: Hoboken, NY, USA, 1984. [Google Scholar]
- Mills, I.M. Vibration-Rotation Structure in Asymmetric- and Symmetric-Top Molecules. In Molecular Spectroscopy: Modern Research; Rao, K.N., Mathews, C.W., Eds.; Academic Press: New York, NY, USA, 1972; Chapter 3.2; pp. 115–140. [Google Scholar]
- Puzzarini, C.; Heckert, J.; Gauss, J. The accuracy of rotational constants predicted by high-level quantum-chemical calculations. I. molecules containing first-row atoms. J. Chem. Phys. 2008, 128, 194108. [Google Scholar] [CrossRef]
- Alessandrini, S.; Gauss, J.; Puzzarini, C. Accuracy of Rotational Parameters Predicted by High-Level Quantum-Chemical Calculations: Case Study of Sulfur-Containing Molecules of Astrochemical Interest. J. Chem. Theory Comput. 2018, 14, 5360–5371. [Google Scholar] [CrossRef] [Green Version]
- Puzzarini, C.; Stanton, J.F.; Gauss, J. Quantum-chemical calculation of spectroscopic parameters for rotational spectroscopy. Int. Rev. Phys. Chem. 2010, 29, 273–367. [Google Scholar] [CrossRef]
- Papajak, E.; Leverentz, H.R.; Zheng, J.; Truhlar, D.G. Efficient Diffuse Basis Sets: Cc-pVxZ+ and maug-cc-pVxZ. J. Chem. Theo. Computat. 2009, 5, 1197–1202. [Google Scholar] [CrossRef]
- Fornaro, T.; Biczysko, M.; Bloino, J.; Barone, V. Reliable vibrational wavenumbers for C=O N–H stretchings of isolated and hydrogen-bonded nucleic acid bases. Phys. Chem. Chem. Phys. 2016, 18, 8479–8490. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Feller, D. The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water. J. Chem. Phys. 1993, 98, 7059–7071. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Tajti, A.; Szalay, P.G.; Császár, A.G.; Kállay, M.; Gauss, J.; Valeev, E.F.; Flowers, B.A.; Vázquez, J.; Stanton, J.F. HEAT: High accuracy extrapolated ab initio thermochemistry. J. Chem. Phys. 2004, 121, 11599–11613. [Google Scholar] [CrossRef] [Green Version]
- Noga, J.; Bartlett, R.J. The Full CCSDT Model for Molecular Electronic Structure. J. Chem. Phys. 1987, 86, 7041–7050. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Schaefer, H.F., III. A New Implementation of the Full CCSDT Model for Molecular Electronic-Structure. Chem. Phys. Lett. 1988, 152, 382–386. [Google Scholar] [CrossRef]
- Watts, J.D.; Bartlett, R.J. The Coupled-Cluster Single, Double, and Triple Excitation Model for Open-Shell Single Reference Functions. J. Chem. Phys. 1990, 93, 6104–6105. [Google Scholar] [CrossRef]
- Bomble, Y.J.; Stanton, J.F.; Kállay, M.; Gauss, J. Coupled-cluster methods including noniterative corrections for quadruple excitations. J. Chem. Phys. 2005, 123, 054101. [Google Scholar] [CrossRef]
- Kállay, M.; Gauss, J. Approximate treatment of higher excitations in coupled-cluster theory. J. Chem. Phys. 2005, 123, 214105. [Google Scholar] [CrossRef]
- Kállay, M.; Gauss, J. Approximate treatment of higher excitations in coupled-cluster theory. II. Extension to general single-determinant reference functions and improved approaches for the canonical Hartree–Fock case. J. Chem. Phys. 2008, 129, 144101. [Google Scholar] [CrossRef]
- Sellers, H.; Pulay, P. The adiabatic correction to molecular potential surfaces in the SCF approximation. Chem. Phys. Lett. 1984, 103, 463. [Google Scholar] [CrossRef]
- Handy, N.C.; Yamaguchi, Y.; Schaefer, H.F. The diagonal correction to the Born–Oppenheimer approximation: Its effect on the singlet–triplet splitting of CH2 and other molecular effects. J. Chem. Phys. 1986, 84, 4481. [Google Scholar] [CrossRef]
- Handy, N.C.; Lee, A.M. The adiabatic approximation. Chem. Phys. Lett. 1996, 252, 425. [Google Scholar] [CrossRef]
- Kutzelnigg, W. The adiabatic approximation I. The physical background of the Born-Handy ansatz. Mol. Phys. 1997, 90, 909. [Google Scholar] [CrossRef]
- Cowan, R.D.; Griffin, M. Approximate relativistic corrections to atomic radial wave functions. J. Opt. Soc. Am. 1976, 66, 1010. [Google Scholar] [CrossRef]
- Martin, R.L. All-electron relativistic calculations on silver hydride. An investigation of the Cowan-Griffin operator in a molecular species. J. Phys. Chem. 1983, 87, 750. [Google Scholar] [CrossRef]
- Heckert, M.; Kállay, M.; Tew, D.P.; Klopper, W.; Gauss, J. Basis-Set Extrapolation Techniques for the Accurate Calculation of Molecular Equilibrium Geometries Using Coupled-Cluster Theory. J. Chem. Phys. 2006, 125, 044108. [Google Scholar] [CrossRef] [PubMed]
- Heckert, M.; Kállay, M.; Gauss, J. Molecular Equilibrium Geometries Based on Coupled-Cluster Calculations Including Quadruple Excitations. Mol. Phys. 2005, 103, 2109. [Google Scholar] [CrossRef]
- Puzzarini, C.; Barone, V. Extending the molecular size in accurate quantum-chemical calculations: The equilibrium structure and spectroscopic properties of uracil. Phys. Chem. Chem. Phys. 2011, 13, 7189–7197. [Google Scholar] [CrossRef]
- Puzzarini, C.; Biczysko, M.; Barone, V.; Peña, I.; Cabezas, C.; Alonso, J.L. Accurate molecular structure and spectroscopic properties of nucleobases: A combined computational–microwave investigation of 2-thiouracil as a case study. Phys. Chem. Chem. Phys. 2013, 15, 16965–16975. [Google Scholar] [CrossRef] [Green Version]
- Puzzarini, C.; Biczysko, M.; Barone, V.; Largo, L.; Peña, I.; Cabezas, C.; Alonso, J.L. Accurate Characterization of the Peptide Linkage in the Gas Phase: A Joint Quantum-Chemical and Rotational Spectroscopy Study of the Glycine Dipeptide Analogue. J. Phys. Chem. Lett. 2014, 5, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Georgievskii, Y.; Miller, J.A.; Burke, M.P.; Klippenstein, S.J. Reformulation and solution of the master equation for multiple-well chemical reactions. J. Phys. Chem. A 2013, 117, 12146–12154. [Google Scholar] [CrossRef]
- Pechukas, P.; Light, J.C. On detailed balancing and statistical theories of chemical kinetics. J. Chem. Phys. 1965, 42, 3281–3291. [Google Scholar] [CrossRef]
- Chesnavich, W.J. Multiple transition states in unimolecular reactions. J. Chem. Phys. 1986, 84, 2615–2619. [Google Scholar] [CrossRef]
- Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 1930, 35, 1303. [Google Scholar] [CrossRef]
- Vazart, F.; Latouche, C.; Skouteris, D.; Balucani, N.; Barone, V. Cyanomethanimine Isomers in Cold Interstellar Clouds: Insights from Electronic Structure and Kinetic Calculations. Astrophys. J. 2015, 810, 111. [Google Scholar] [CrossRef] [Green Version]
- Skouteris, D.; Balucani, N.; Ceccarelli, C.; Faginas Lago, N.; Codella, C.; Falcinelli, S.; Rosi, M. Interstellar dimethyl ether gas-phase formation: A quantum chemistry and kinetics study. Mon. Not. R. Astron. Soc. 2018, 482, 3567–3575. [Google Scholar] [CrossRef] [Green Version]
- Licari, D.; Tasinato, N.; Spada, L.; Puzzarini, C.; Barone, V. VMS-ROT: A New Module of the Virtual Multifrequency Spectrometer for Simulation, Interpretation, and Fitting of Rotational Spectra. J. Chem. Theory Comput. 2017, 13, 4382–4396. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B2 | CBS+CV a | CBS+CV+fT+pQ b | HEAT-Like b | ChS | Harm-ZPE c | Anharm-ZPE c | d | e | |
|---|---|---|---|---|---|---|---|---|---|
| NH + C2H5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| M1 | −345.16 | −352.10 | −351.92 | −352.09 | −352.82 | 28.40 | 28.13 | −323.97 | −311 |
| (−352.01) | (−351.83) | (−352.00) | (-323) | ||||||
| M2 | −342.24 | −349.43 | −349.27 | −349.44 | −350.28 | 28.94 | 28.59 | −320.84 | −308 |
| (−349.33) | (−349.18) | (−349.35) | (−320) | ||||||
| M3 | −382.90 | −388.91 | - | - | −390.12 | 31.07 | 30.08 | −358.83 | −340 |
| (−388.89) | (−357) | ||||||||
| M4 | −336.75 | −345.98 | - | - | −347.39 | 28.74 | 28.30 | −317.68 | −301 |
| (−345.71) | - | ||||||||
| TS1 | −338.10 | −345.27 | −345.11 | −345.28 | −346.14 | 28.51 | 28.17 | −317.10 | −304 |
| (−345.20) | (−345.04) | (−345.21) | (−316) | ||||||
| TS2 | −223.85 | −226.32 | −229.11 | −229.22 | −227.56 | 18.47 | 18.43 | −207.89 | −197 |
| (−226.58) | (−229.37) | (−229.48) | (−211) | ||||||
| TS3 | −195.51 | −202.17 | −203.93 | −204.09 | −202.88 | 9.31 | 8.85 | −193.32 | −180 |
| (−202.41) | (−204.18) | (−204.34) | (−197) | ||||||
| TS4 | −192.92 | −199.72 | −201.47 | −201.62 | −200.42 | 9.48 | 9.07 | −190.65 | −177 |
| (−199.95) | (−201.70) | (−201.85) | (−194) | ||||||
| TS-isom | −94.55 | −98.47 | −98.17 | −98.40 | −100.55 | −4.13 | −4.00 | −102.47 | −84 |
| (−98.81) | (−98.51) | (−98.74) | (−101) | ||||||
| TS5 | −190.43 | −197.33 | - | - | −199.01 | 18.79 | 18.19 | −179.14 | −165 |
| (−197.27) | - | ||||||||
| TS6 | −176.42 | −182.49 | - | - | −184.40 | 17.70 | 16.87 | −165.62 | −146 |
| (−182.51) | - | ||||||||
| TS7 | −236.01 | −237.83 | - | - | −238.77 | 20.67 | 20.63 | −217.20 | −207 |
| (−238.27) | - | ||||||||
| CH2NH + CH3 | −245.85 | −251.18 | −251.36 | −251.34 | −253.69 | 7.24 | 7.47 | -243.71 | -230 |
| (−251.38) | (−251.57) | (−251.55) | (−243) | ||||||
| E−CH3CHNH + H | −213.82 | −222.30 | −221.99 | −221.92 | −224.10 | 4.00 | 4.14 | −218.16 | −202 |
| (−222.65) | (−222.34) | (−222.27) | (−217) | ||||||
| Z−CH3CHNH + H | −210.94 | −219.52 | −219.22 | −219.13 | −221.31 | 4.06 | 4.20 | −215.32 | −199 |
| (−219.85) | (−219.55) | (−219.46) | (−214) | ||||||
| C2H4 + NH2 | −242.67 | −248.70 | - | - | −250.94 | 8.26 | 8.64 | -240.06 | −228 |
| (−248.78) | (−241) |
| T (K) | CH3 + CH2NH | E−CH3CHNH | Z−CH3CHNH | C2H4 + NH2 |
|---|---|---|---|---|
| 10 | 1.13 × 10−10 | 8.87 × 10−12 | 6.29 × 10−12 | 7.19 × 10−13 |
| 20 | 1.28 × 10−10 | 8.45 × 10−12 | 6.35 × 10−12 | 5.98 × 10−13 |
| 30 | 1.38 × 10−10 | 8.64 × 10−12 | 6.61 × 10−12 | 5.90 × 10−13 |
| 40 | 1.45 × 10−10 | 8.91 × 10−12 | 6.85 × 10−12 | 6.00 × 10−13 |
| 50 | 1.50 × 10−10 | 9.18 × 10−12 | 7.08 × 10−12 | 6.14 × 10−13 |
| 60 | 1.55 × 10−10 | 9.41 × 10−12 | 7.27 × 10−12 | 6.27 × 10−13 |
| 70 | 1.59 × 10−10 | 9.67 × 10−12 | 7.47 × 10−12 | 6.45 × 10−13 |
| 80 | 1.63 × 10−10 | 9.90 × 10−12 | 7.64 × 10−12 | 6.61 × 10−13 |
| 90 | 1.66 × 10−10 | 1.01 × 10−11 | 7.80 × 10−12 | 6.76 × 10−13 |
| 100 | 1.69 × 10−10 | 1.03 × 10−11 | 7.95 × 10−12 | 6.90 × 10−13 |
| 110 | 1.71 × 10−10 | 1.05 × 10−11 | 8.08 × 10−12 | 7.03 × 10−13 |
| 120 | 1.74 × 10−10 | 1.07 × 10−11 | 8.21 × 10−12 | 7.16 × 10−13 |
| 130 | 1.76 × 10−10 | 1.08 × 10−11 | 8.33 × 10−12 | 7.28 × 10−13 |
| 140 | 1.78 × 10−10 | 1.10 × 10−11 | 8.44 × 10−12 | 7.39 × 10−13 |
| 150 | 1.80 × 10−10 | 1.11 × 10−11 | 8.55 × 10−12 | 7.50 × 10−13 |
| 160 | 1.82 × 10−10 | 1.12 × 10−11 | 8.65 × 10−12 | 7.60 × 10−13 |
| 170 | 1.84 × 10−10 | 1.14 × 10−11 | 8.75 × 10−12 | 7.70 × 10−13 |
| 180 | 1.86 × 10−10 | 1.15 × 10−11 | 8.84 × 10−12 | 7.80 × 10−13 |
| 190 | 1.87 × 10−10 | 1.16 × 10−11 | 8.93 × 10−12 | 7.90 × 10−13 |
| 200 | 1.89 × 10−10 | 1.18 × 10−11 | 9.02 × 10−12 | 7.99 × 10−13 |
| 210 | 1.90 × 10−10 | 1.19 × 10−11 | 9.11 × 10−12 | 8.09 × 10−13 |
| 220 | 1.92 × 10−10 | 1.20 × 10−11 | 9.19 × 10−12 | 8.18 × 10−13 |
| 230 | 1.93 × 10−10 | 1.21 × 10−11 | 9.27 × 10−12 | 8.26 × 10−13 |
| 240 | 1.94 × 10−10 | 1.22 × 10−11 | 9.35 × 10−12 | 8.35 × 10−13 |
| 250 | 1.96 × 10−10 | 1.23 × 10−11 | 9.42 × 10−12 | 8.44 × 10−13 |
| 260 | 1.97 × 10−10 | 1.24 × 10−11 | 9.49 × 10−12 | 8.52 × 10−13 |
| 270 | 1.98 × 10−10 | 1.25 × 10−11 | 9.57 × 10−12 | 8.61 × 10−13 |
| 280 | 1.99 × 10−10 | 1.26 × 10−11 | 9.64 × 10−12 | 8.69 × 10−13 |
| 290 | 2.00 × 10−10 | 1.27 × 10−11 | 9.71 × 10−12 | 8.78 × 10−13 |
| 300 | 2.01 × 10−10 | 1.28 × 10−11 | 9.77 × 10−12 | 8.86 × 10−13 |
| Branching Ratios | CH3 + CH2NH | E−CH3CHNH + H | Z−CH3CHNH + H | C2H4 + NH2 |
|---|---|---|---|---|
| 10 K | 87.7% | 6.9% | 4.9% | 0.6% |
| 100 K | 89.9% | 5.5% | 4.2% | 0.4% |
| 300 K | 89.6% | 5.7% | 4.3% | 0.4% |
| E-CH3CHNH | Z-CH3CHNH | |||||
|---|---|---|---|---|---|---|
| Theory | Experiment | Theory | Experiment | |||
| Best Estimates | B2 | Best Estimates | B2 | |||
| 53,178.26 | 53,398.97 | 53,120.561(30) | 50,002.63 | 50,288.21 | 49,964.87(93) | |
| 9780.14 | 9744.02 | 9782.7720(47) | 9831.98 | 9781.22 | 9832.4823(96) | |
| 8702.82 | 8679.22 | 8697.0263(46) | 8652.81 | 8621.21 | 8646.0305(94) | |
| 6.48 | 6.39 | 6.4641(49) | 6.99 | 6.96 | 6.938(13) | |
| 0.568 | 0.575 | 0.5763(34) | 0.468 | 0.480 | 0.468 | |
| −0.0165 | −0.0165 | −0.01403(21) | −0.0163 | −0.0182 | −0.01219(23) | |
| 1.09 | 1.06 | 1.1033(19) | 1.25 | 1.24 | 1.2657(65) | |
| −0.0535 | −0.0518 | −0.06709(59) | −0.0522 | −0.0464 | −0.0642(19) | |
| 563.1 | 488.8 | 566.37(20) | 523.3 | 446.4 | 517.41(33) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baiano, C.; Lupi, J.; Tasinato, N.; Puzzarini, C.; Barone, V. The Role of State-of-the-Art Quantum-Chemical Calculations in Astrochemistry: Formation Route and Spectroscopy of Ethanimine as a Paradigmatic Case. Molecules 2020, 25, 2873. https://doi.org/10.3390/molecules25122873
Baiano C, Lupi J, Tasinato N, Puzzarini C, Barone V. The Role of State-of-the-Art Quantum-Chemical Calculations in Astrochemistry: Formation Route and Spectroscopy of Ethanimine as a Paradigmatic Case. Molecules. 2020; 25(12):2873. https://doi.org/10.3390/molecules25122873
Chicago/Turabian StyleBaiano, Carmen, Jacopo Lupi, Nicola Tasinato, Cristina Puzzarini, and Vincenzo Barone. 2020. "The Role of State-of-the-Art Quantum-Chemical Calculations in Astrochemistry: Formation Route and Spectroscopy of Ethanimine as a Paradigmatic Case" Molecules 25, no. 12: 2873. https://doi.org/10.3390/molecules25122873
APA StyleBaiano, C., Lupi, J., Tasinato, N., Puzzarini, C., & Barone, V. (2020). The Role of State-of-the-Art Quantum-Chemical Calculations in Astrochemistry: Formation Route and Spectroscopy of Ethanimine as a Paradigmatic Case. Molecules, 25(12), 2873. https://doi.org/10.3390/molecules25122873