
Reactions of Dihaloboranes with Electron-Rich 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes



Abstract
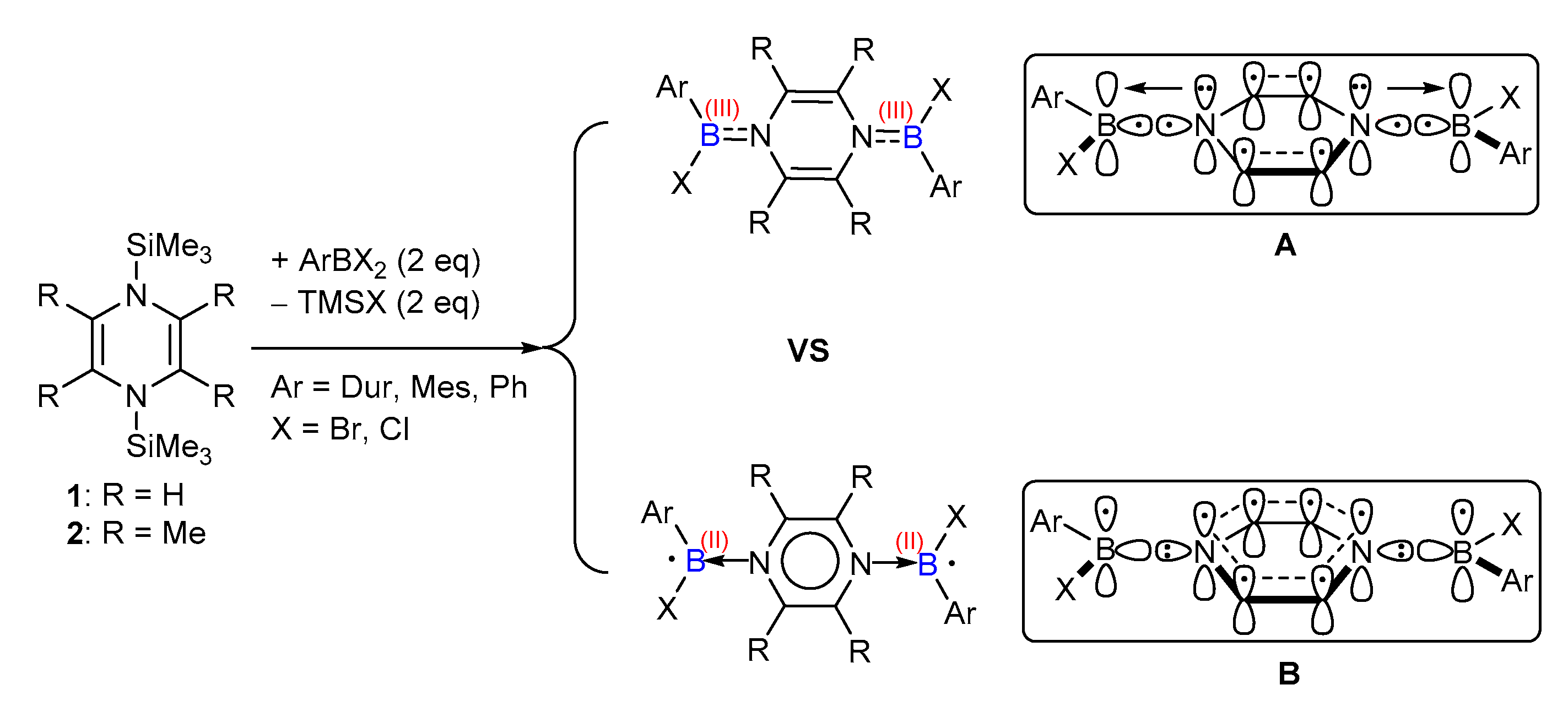
:1. Introduction
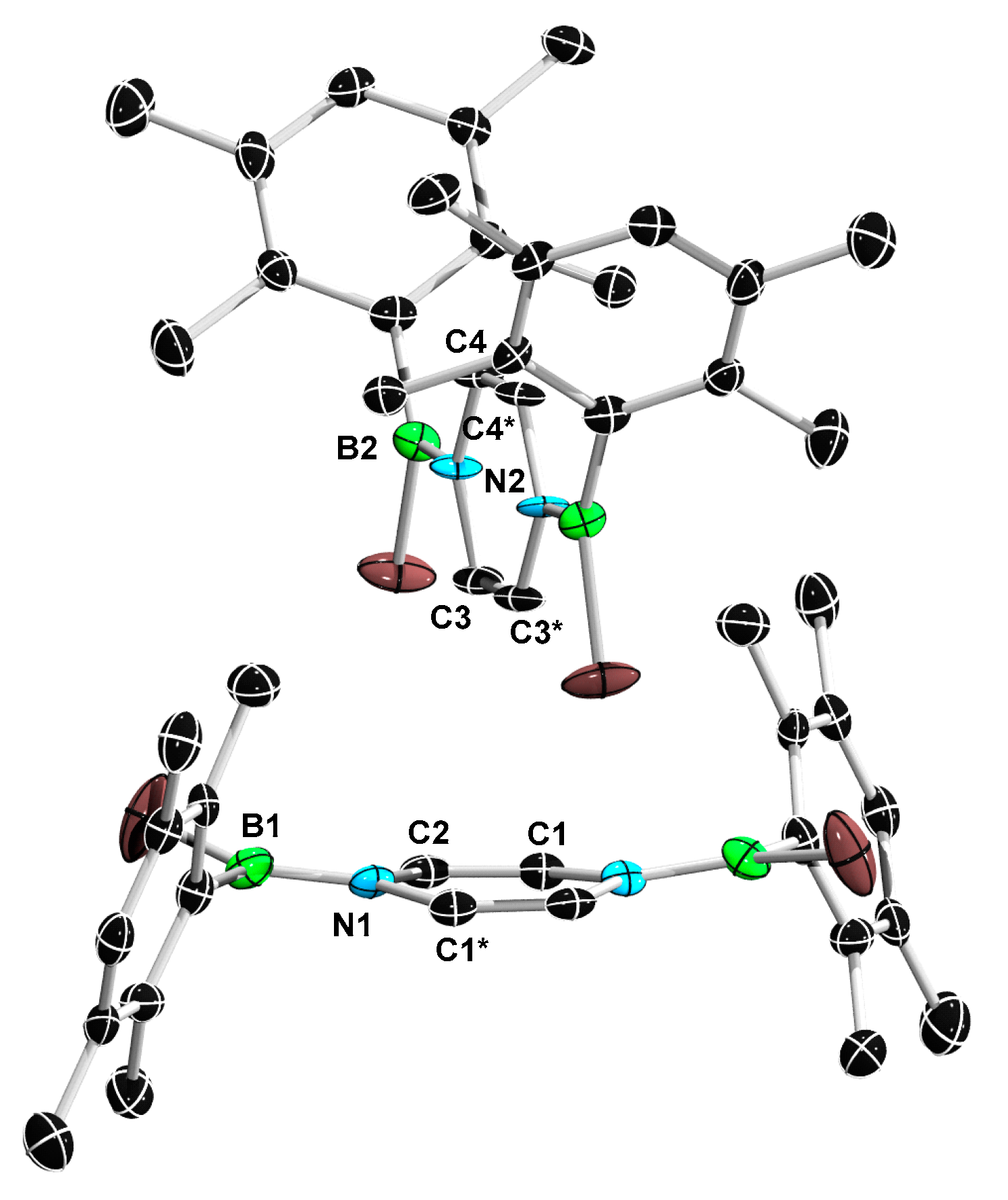
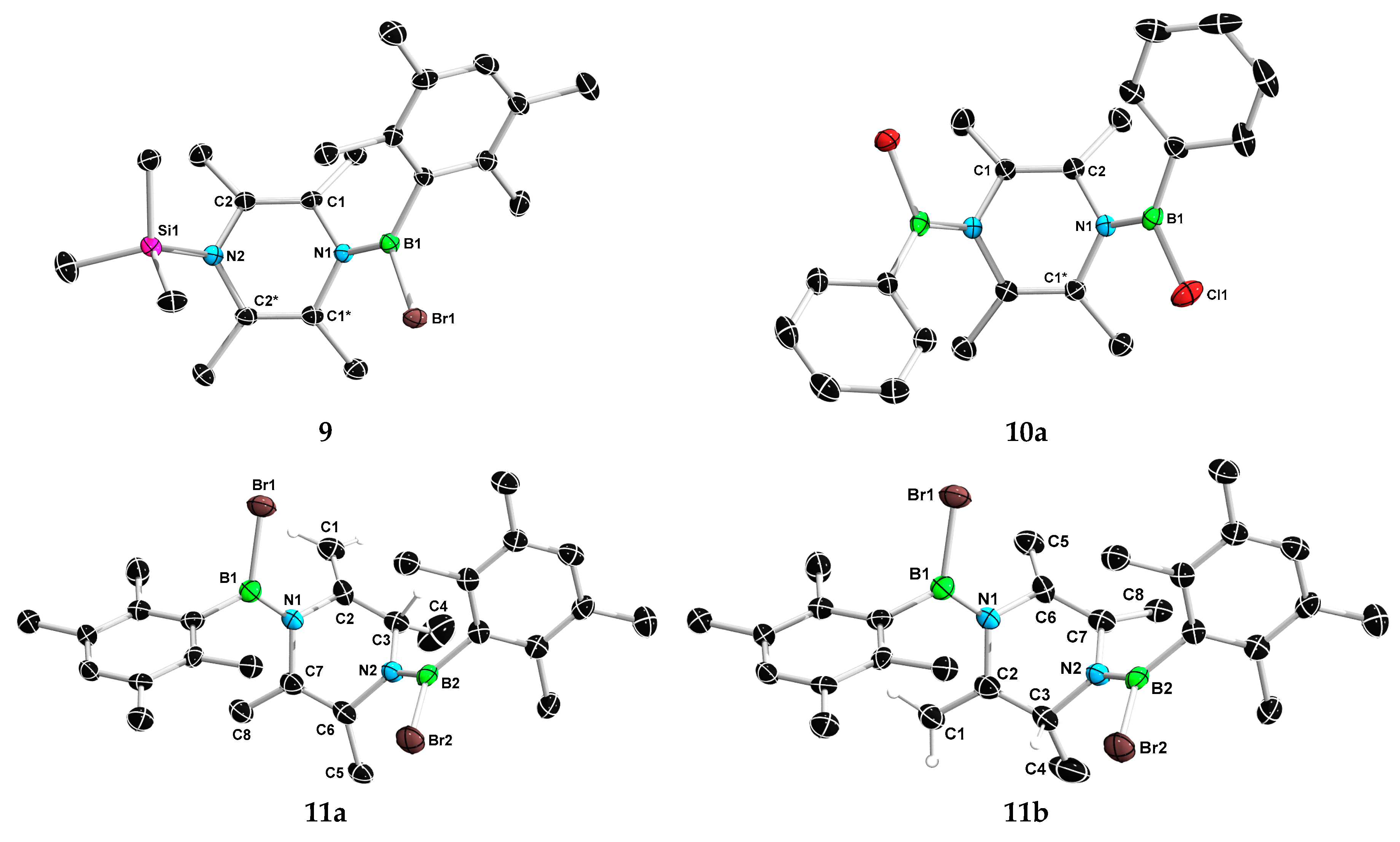
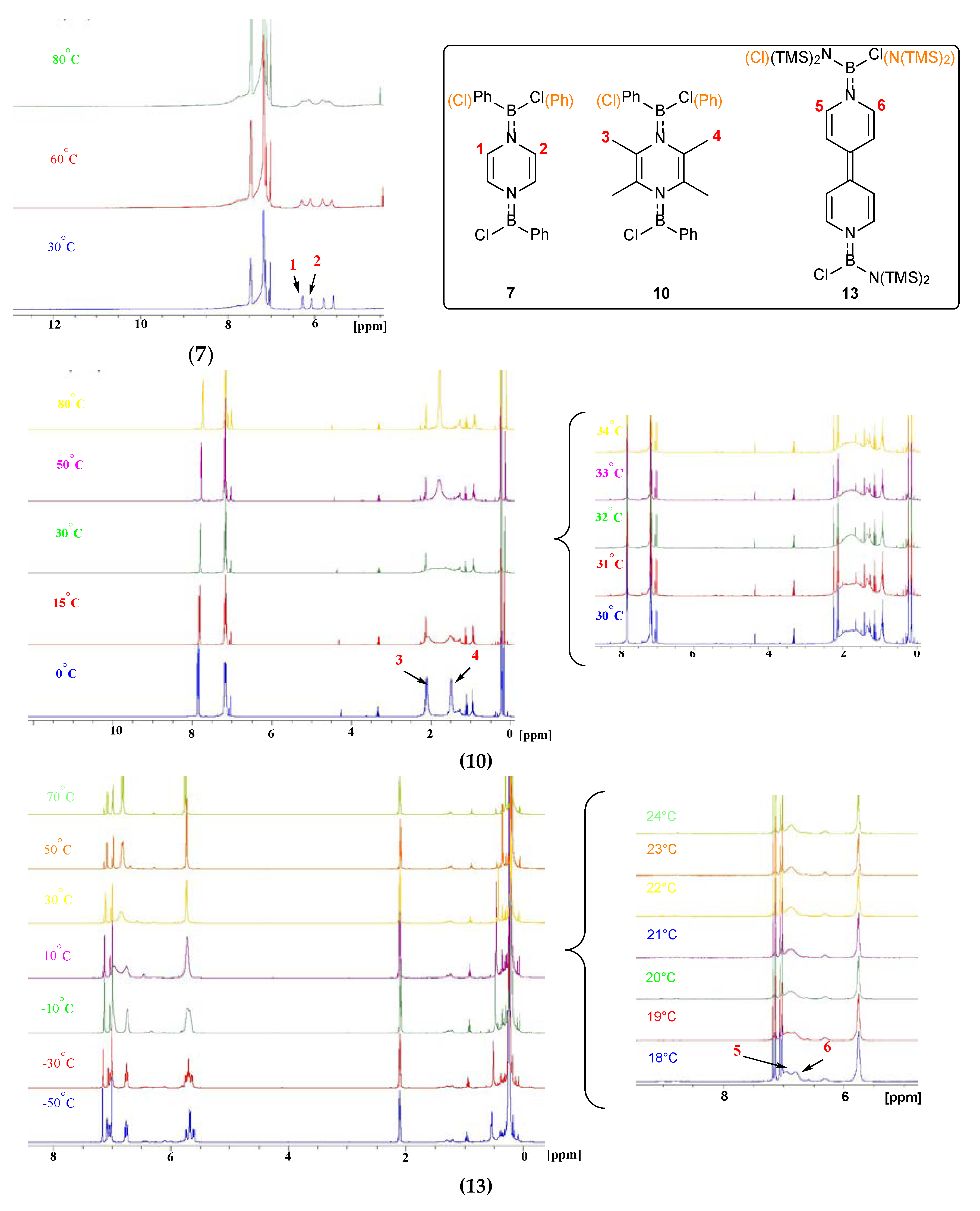
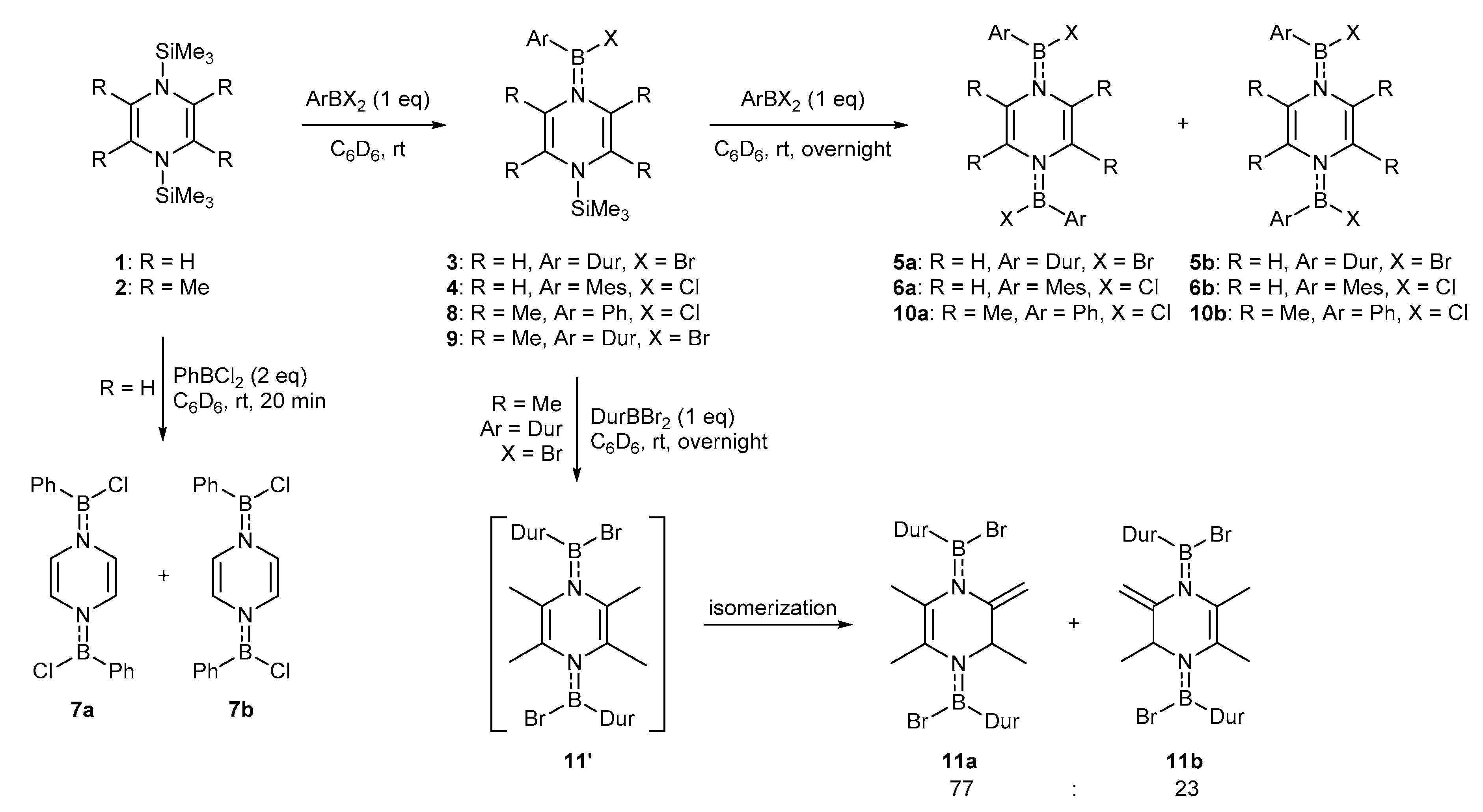
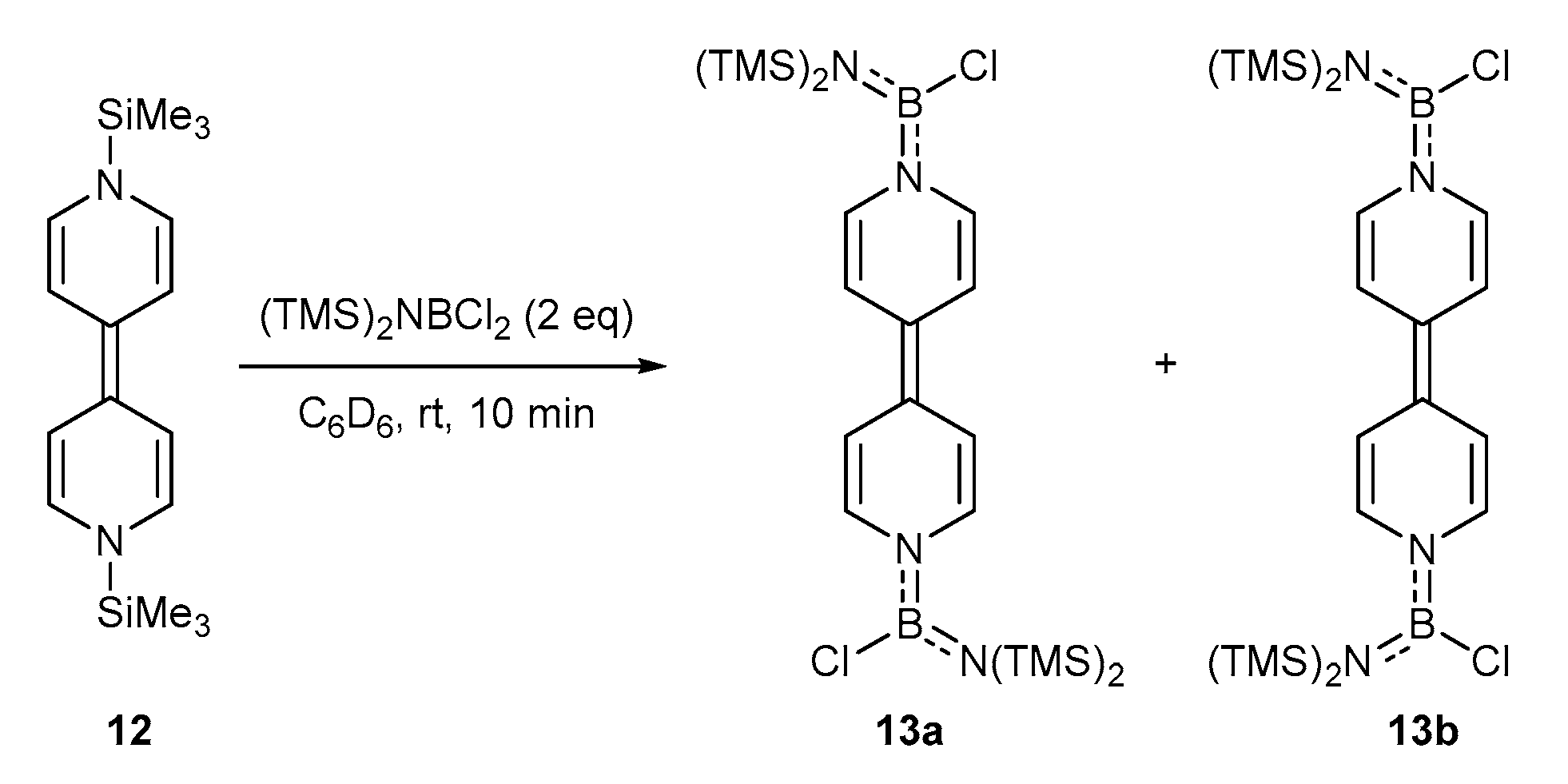
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 3 and 4:
3.3. Synthesis of 5–7
3.4. Synthesis of 8 and 9
3.5. Synthesis of 10
3.6. Synthesis of 11a and 11b
3.7. Synthesis of 13a and 13b
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krasowska, M.; Bettinger, H.F. Reactivity of borylenes toward ethyne, ethene, and methane. J. Am. Chem. Soc. 2012, 134, 17094–17103. [Google Scholar] [CrossRef] [PubMed]
- Krasowska, M.; Edelmann, M.; Bettinger, H.F. Electronically excited states of borylenes. J. Phys. Chem. A 2016, 120, 6332–6341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Robinson, G.H. Carbene stabilization of highly reactive main-group molecules. Inorg. Chem. 2011, 50, 12326–12337. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, P.; Braunschweig, H.; Damme, A.; Dewhurst, R.D.; Kupfer, T.; Radacki, K.; Wagner, K. Generation of a carbene-stabilized bora-borylene and its insertion into a C–H bond. J. Am. Chem. Soc. 2011, 133, 19044–19047. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, P.; Braunschweig, H.; Kraft, K.; Kupfer, T. Trapping the elusive parent borylene. Angew. Chem. Int. Ed. 2011, 50, 4704–4707. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; Boussonnière, A.; Geib, S.J.; Lacôte, E. The parent borylene: Betwixt and between. Angew. Chem. Int. Ed. 2012, 51, 1602–1605. [Google Scholar] [CrossRef]
- Braunschweig, H.; Claes, C.; Damme, A.; Deißenberger, A.; Dewhurst, R.D.; Hörl, C.; Kramer, T. A facile and selective route to remarkably inert monocyclic NHC-stabilized boriranes. Chem. Commun. 2015, 51, 1627–1630. [Google Scholar] [CrossRef]
- Frey, G.D.; Lavallo, V.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 2007, 316, 439–441. [Google Scholar] [CrossRef] [Green Version]
- Légaré, M.-A.; Bélanger-Chabot, G.; Dewhurst, R.D.; Welz, E.; Krummenacher, I.; Engels, B.; Braunschweig, H. Nitrogen fixation and reduction at boron. Science 2018, 359, 896–900. [Google Scholar] [CrossRef] [Green Version]
- Légaré, M.-A.; Rang, M.; Bélanger-Chabot, G.; Schweizer, J.I.; Krummenacher, I.; Bertermann, R.; Arrowsmith, M.; Holthausen, M.C.; Braunschweig, H. The reductive coupling of dinitrogen. Science 2019, 363, 1329–1332. [Google Scholar] [CrossRef]
- Krasowska, M.; Bettinger, H.F. Ring enlargement of three-membered boron heterocycles upon reaction with organic π systems: Implications for the trapping of borylenes. Chem. Eur. J. 2016, 22, 10661–10670. [Google Scholar] [CrossRef] [PubMed]
- Timms, P.L. Boron-fluorine chemistry. I. Boron monofluoride and some derivatives. J. Am. Chem. Soc. 1967, 89, 1629–1632. [Google Scholar] [CrossRef]
- Timms, P.L. Chemistry of boron and silicon subhalides. Acc. Chem. Res. 1973, 6, 118–123. [Google Scholar] [CrossRef]
- Wang, Y.; Quillian, B.; Wei, P.; Wannere, C.S.; Xie, Y.; King, R.B.; Schaefer, H.F.; Schleyer, P.v.R.; Robinson, G.H. A stable neutral diborene containing a B=B double bond. J. Am. Chem. Soc. 2007, 129, 12412–12413. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, R.; Donnadieu, B.; Celik, M.A.; Frenking, G.; Bertrand, G. Synthesis and characterization of a neutral tricoordinate organoboron isoelectronic with amines. Science 2011, 333, 610–613. [Google Scholar] [CrossRef]
- Woon, E.C.Y.; Tumber, A.; Kawamura, A.; Hillringhaus, L.; Ge, W.; Rose, N.R.; Ma, J.H.Y.; Chan, M.C.; Walport, L.J.; Che, K.H.; et al. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of Jmjc histone demethylases. Angew. Chem. Int. Ed. 2012, 51, 1631–1634. [Google Scholar] [CrossRef]
- Kong, L.; Li, Y.; Ganguly, R.; Vidovic, D.; Kinjo, R. Isolation of a bis(oxazol-2-ylidene)–phenylborylene adduct and its reactivity as a boron-centered nucleophile. Angew. Chem. Int. Ed. 2014, 53, 9280–9283. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, J.; Lin, Z.; Xie, Z. Synthesis and structural characterization of carbene-stabilized carborane-fused azaborolyl radical cation and dicarbollyl-fused azaborole. Organometallics 2016, 35, 2579–2582. [Google Scholar] [CrossRef]
- Lu, W.; Li, Y.; Ganguly, R.; Kinjo, R. Alkene-carbene isomerization induced by borane: Access to an asymmetrical diborene. J. Am. Chem. Soc. 2017, 139, 5047–5050. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, J.; Lin, Z.; Xie, Z. The synthesis and structure of a carbene-stabilized iminocarboranyl-boron(I) compound. Chem. Commun. 2015, 51, 16817–16820. [Google Scholar] [CrossRef]
- Ruiz, D.A.; Melaimi, M.; Bertrand, G. An efficient synthetic route to stable bis(carbene)borylenes [(L1)(L2)BH]. Chem. Commun. 2014, 50, 7837–7839. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Wittenbecher, L.; Boese, R.; Bläser, D. N,N′-bis(2,2-dimethylpropyl)benzimidazolin-2-ylidene: A stable nucleophilic carbene derived from benzimidazole. Chem. Eur. J. 1999, 5, 1931–1935. [Google Scholar] [CrossRef]
- Lavallo, V.; Canac, Y.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Cyclopropenylidenes: From interstellar space to an isolated derivative in the laboratory. Science 2006, 312, 722–724. [Google Scholar] [CrossRef] [Green Version]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Broggi, J.; Terme, T.; Vanelle, P. Organic electron donors as powerful single-electron reducing agents in organic synthesis. Angew. Chem. Int. Ed. 2014, 53, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Eberle, B.; Hubner, O.; Ziesak, A.; Kaifer, E.; Himmel, H.J. What makes a strong organic electron donor (or acceptor)? Chem. Eur. J. 2015, 21, 8578–8590. [Google Scholar] [CrossRef]
- Tsurugi, H.; Saito, T.; Tanahashi, H.; Arnold, J.; Mashima, K. Carbon radical generation by d0 tantalum complexes with α-diimine ligands through ligand-centered redox processes. J. Am. Chem. Soc. 2011, 133, 18673–18683. [Google Scholar] [CrossRef]
- Tsurugi, H.; Tanahashi, H.; Nishiyama, H.; Fegler, W.; Saito, T.; Sauer, A.; Okuda, J.; Mashima, K. Salt-free reducing reagent of bis(trimethylsilyl)cyclohexadiene mediates multielectron reduction of chloride complexes of W(VI) and W(IV). J. Am. Chem. Soc. 2013, 135, 5986–5989. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, H.; Tanahashi, H.; Kawakita, K.; Tsurugi, H.; Mashima, K. 1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as strong salt-free reductants for generating low-valent early transition metals with electron-donating ligands. J. Am. Chem. Soc. 2014, 136, 5161–5170. [Google Scholar] [CrossRef]
- Tsurugi, H.; Mashima, K. A new protocol to generate catalytically active species of group 4–6 metals by organosilicon-based salt-free reductants. Chem. Eur. J. 2019, 25, 913–919. [Google Scholar] [CrossRef]
- Arteaga-Muller, R.; Tsurugi, H.; Saito, T.; Yanagawa, M.; Oda, S.; Mashima, K. New tantalum ligand-free catalyst system for highly selective trimerization of ethylene affording 1-hexene: New evidence of a metallacycle mechanism. J. Am. Chem. Soc. 2009, 131, 5370–5371. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishiyama, H.; Kawakita, K.; Nechayev, M.; Kriegel, B.; Tsurugi, H.; Arnold, J.; Mashima, K. Reduction of (tBuN=)NbCl3(py)2 in a salt-free manner for generating Nb(IV) dinuclear complexes and their reactivity toward benzo[c]cinnoline. Inorg. Chem. 2015, 54, 6004–6009. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, H.; Ikeda, H.; Tsurugi, H.; Mashima, K. Synthesis and characterization of paramagnetic tungsten imido complexes bearing α-diimine ligands. Inorg. Chem. 2016, 55, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Weyenberg, D.R.; Toporcer, L.H. The synthesis of 3,6-disilyl-1,4-cyclohexadienes by the trapping of benzene anion-radicals. J. Am. Chem. Soc. 1962, 84, 2843–2844. [Google Scholar] [CrossRef]
- Braunschweig, H.; Ye, Q.; Radacki, K. High yield synthesis of a neutral and carbonyl-rich terminal arylborylene complex. Chem. Commun. 2012, 48, 2701–2703. [Google Scholar] [CrossRef]
- Cowherd, F.G.; Von Rosenberg, J.L. Mechanism of iron pentacarbonyl-catalyzed 1,3-hydrogen shifts. J. Am. Chem. Soc. 1969, 91, 2157–2158. [Google Scholar] [CrossRef]
- Hvistendahl, G.; Williams, D.H. Energy barrier to symmetry-forbidden 1,3-hydrogen shifts in simple oxonium ions. Metastable peaks from fast dissociations. J. Am. Chem. Soc. 1975, 97, 3097–3101. [Google Scholar] [CrossRef]
- Laguerre, M.; Dunogues, J.; Calas, R.; Duffaut, N. Silylation d′hydrocarbures mono-aromatiques mono- ou disubstitues. J. Organomet. Chem. 1976, 112, 49–59. [Google Scholar] [CrossRef]
- Lis, A.V.; Gostevskii, B.A.; Albanov, A.I.; Yarosh, N.O.; Rakhlin, V.I. Synthesis of volatile bis[bis(trimethylsilyl)amide]-substituted boron derivatives. Russ. J. Gen. Chem. 2017, 87, 353–356. [Google Scholar] [CrossRef]
- Brown, C.; Cragg, R.H.; Miller, T.J.; Smith, D.O.N. Organoboron compounds: Xxii. A 13C NMR study of some dialkylaminophenylboranes. J. Organomet. Chem. 1983, 244, 209–215. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Tc | △ν | △G≠ |
|---|---|---|---|
| 7 | >80 °C (353 K) | 85.0 Hz | >71.56 kJ/mol |
| 10 | 32 °C (305 K) | 243.5 Hz | 58.79 kJ/mol |
| 13 | 20 °C (293 K) | 96.0 Hz | 58.65 kJ/mol |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, L.; Zhang, X.; Ming, W.; Su, S.; Chang, X.; Ye, Q. Reactions of Dihaloboranes with Electron-Rich 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes. Molecules 2020, 25, 2875. https://doi.org/10.3390/molecules25122875
Ma L, Zhang X, Ming W, Su S, Chang X, Ye Q. Reactions of Dihaloboranes with Electron-Rich 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes. Molecules. 2020; 25(12):2875. https://doi.org/10.3390/molecules25122875
Chicago/Turabian StyleMa, Li, Xiaolin Zhang, Wenbo Ming, Shengxin Su, Xiaoyong Chang, and Qing Ye. 2020. "Reactions of Dihaloboranes with Electron-Rich 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes" Molecules 25, no. 12: 2875. https://doi.org/10.3390/molecules25122875
APA StyleMa, L., Zhang, X., Ming, W., Su, S., Chang, X., & Ye, Q. (2020). Reactions of Dihaloboranes with Electron-Rich 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes. Molecules, 25(12), 2875. https://doi.org/10.3390/molecules25122875