
Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts




Abstract
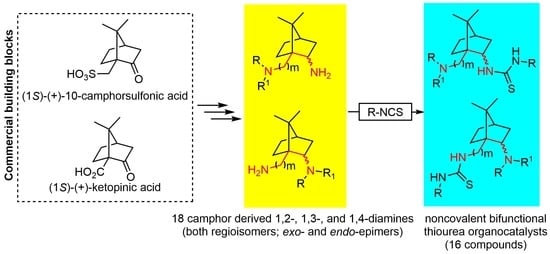
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Camphor-Derived 1,2-Diamines
2.2. Synthesis of Camphor Derived 1,3-Diamines
2.3. Synthesis of Camphor Derived 1,4-Diamines
2.4. Synthesis of Noncovalent Thiourea Organocatalysts 48–63
2.5. Structure Determination
2.6. Performance of Noncovalent Thiourea Organocatalysts 48–63 in 1,4-Addition of 1,3-Dicarbonyl Compounds to trans-β-Nitrostyrene
3. Materials and Methods
3.1. Materials and General Methods
3.2. General Procedures
3.2.1. General Procedure 1. Synthesis of Free Diamines by Deprotection of N-Boc-Amines
3.2.2. General Procedure 2. Synthesis of Oximes
3.2.3. General Procedure 3. Synthesis of Tertiary Amines (Pyrrolidines) by Cyclative Bis-Alkylation of Primary Amines
3.2.4. General Procedure 4. Synthesis of Primary Amines by Reduction of Oximes with Sodium
3.2.5. General Procedure 5. Synthesis of Primary Amines by Hydrogenation of Oximes in the Presence of Raney-Ni Catalyst
3.2.6. General Procedure 6. Synthesis of Thiourea Derivatives
3.2.7. General Procedure 7. Synthesis of N-Boc Protected Primary Amines
3.2.8. General Procedure 8. Synthesis of Tertiary Amines by Amination of 10-Iodocamphor (18)
3.2.9. General Procedure 10. Testing the Catalytic Activity of Thiourea Derivatives in 1,4-Additions of 1,3-Dicarbonyl Compounds to trans-β-Nitrostyrene
3.3. General Procedures
3.3.1. Synthesis of 1-[3,5-Bis(trifluoromethyl)phenyl]-3-[(1S,2R,4R)-7,7-dimethyl-1-(pyrrolidin-1-yl)-bicyclo[2.2.1]heptan-2-yl]thiourea (48)
3.3.2. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1S,2S,4R)-7,7-dimethyl-1-(pyrrolidin-1-yl)-bicyclo[2.2.1]heptan-2-yl)thiourea (49)
3.3.3. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1S,2R,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)-bicyclo[2.2.1]heptan-1-yl)thiourea (50)
3.3.4. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1S,2S,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)-bicyclo[2.2.1]heptan-1-yl)thiourea (51)
3.3.5. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1R,2S,4R)-7,7-dimethyl-1-(pyrrolidin-1-yl-methyl)bicyclo[2.2.1]heptan-2-yl)thiourea (52)
3.3.6. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1R,2R,4R)-7,7-dimethyl-1-(pyrrolidin-1-yl-methyl)bicyclo[2.2.1]heptan-2-yl)thiourea (53)
3.3.7. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1R,2S,4R)-7,7-dimethyl-1-(piperidin-1-yl-methyl)bicyclo[2.2.1]heptan-2-yl)thiourea (54)
3.3.8. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1R,2S,4R)-7,7-dimethyl-1-(morpholino-methyl)bicyclo[2.2.1]heptan-2-yl)thiourea (55)
3.3.9. Synthesis of 1-(3,5-Bis(trifluoromethyl)phenyl)-3-((1R,2S,4R)-1-((dimethylamino)methyl)-7,7-dimethylbicyclo[2.2.1]heptan-2-yl)thiourea (56)
3.3.10. Synthesis of 1-(tert-butyl)-3-((1R,2S,4R)-7,7-dimethyl-1-(pyrrolidin-1-ylmethyl)bicyclo-[2.2.1]heptan-2-yl)thiourea (57)
3.3.11. Synthesis of 1-(Adamantan-1-yl)-3-((1R,2S,4R)-7,7-dimethyl-1-(pyrrolidin-1-ylmethyl)-bicyclo[2.2.1]heptan-2-yl)thiourea (58)
3.3.12. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-(((1R,2S,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)bicyclo[2.2.1]heptan-1-yl)methyl)thiourea (59)
3.3.13. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-(((1R,2R,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)bicyclo[2.2.1]heptan-1-yl)methyl)thiourea (60)
3.3.14. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1S,2S,4R)-7,7-dimethyl-1-(2-(pyrrolidin-1-yl)ethyl)bicyclo[2.2.1]heptan-2-yl)thiourea (61)
3.3.15. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-(2-((1S,2R,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)bicyclo[2.2.1]heptan-1-yl)ethyl)thiourea (62)
3.3.16. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-(2-((1S,2S,4R)-7,7-dimethyl-2-(pyrrolidin-1-yl)bicyclo[2.2.1]heptan-1-yl)ethyl)thiourea (63)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Meerwein, H. Über den Reaktionsmechanismus der Umwandlung von Borneol in Camphen; [Dritte Mitteilung über Pinakolinumlagerungen.]. Justus Liebigs Ann. Chem. 1914, 405, 129–175. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.R. Wagner-Meerwein rearrangements. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; Volume 3, pp. 705–719. [Google Scholar]
- Money, T. Remote functionalization of camphor: Application to natural product synthesis. Org. Synth. Theory Appl. 1996, 3, 1–83. [Google Scholar]
- Money, T. Camphor: A chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985, 2, 253–289. [Google Scholar] [CrossRef] [PubMed]
- Holton, R.A.; Somoza, C.; Kim, H.-B.; Liang, F.; Biediger, R.J.; Boatman, P.D.; Shindo, M.; Smith, C.C.; Kim, S.; Nadizadeh, H.; et al. The Total Synthesis of Paclitaxel Starting with Camphor. ACS Symp. Ser. 1995, 583, 288–301. [Google Scholar]
- Nicolaou, K.C.; Yang, Z.; Liu, J.J.; Ueno, H.; Nantermet, P.G.; Guy, R.K.; Claiborne, C.F.; Renaud, J.; Couladouros, E.A.; Paulvannan, K.; et al. Total synthesis of taxol. Nature 1994, 367, 630–634. [Google Scholar] [CrossRef]
- Oppolzer, W. Camphor derivatives as chiral auxiliaries in asymmetric synthesis. Tetrahedron 1987, 43, 1969–2004. [Google Scholar] [CrossRef]
- Oppolzer, W. Camphor as a natural source of chirality in asymmetric synthesis. Pure Appl. Chem. 1990, 62, 1241–1250. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Nugent, W.A. MIB: An advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem. Commun. 1999, 1369–1370. [Google Scholar] [CrossRef]
- Stereoselective Organocatalysis; Rios, R. (Ed.) John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Jakab, G.; Schreiner, P.R. Brønsted Acids: Chiral (Thio)urea Derivatives In Comprehensive Enantioselective Organocatalysis; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 315–342. [Google Scholar]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis–Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-derived heterocycles syntheses and potential applications. Targets Heterocycl. Syst. 2015, 19, 62–100. [Google Scholar]
- Rajaram, S.; Sigman, M.S. Design of Hydrogen Bond Catalysts Based on a Modular Oxazoline Template: Application to an Enantioselective Hetero Diels−Alder Reaction. Org. Lett. 2005, 7, 5473–5475. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Lerner, R.A.; Barbas III, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Sigman, M.S.; Vachal, P.; Jacobsen, E.N. A general catalyst for the asymmetric Strecker reaction. Angew. Chem. Int. Ed. 2000, 39, 1279–1281. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kuan, H.-H.; Chou, T.-Y.; Chen, K. Novel Prolinamide-Camphor-Containing Organocatalysts for Direct Asymmetric Michael Addition of Unmodified Aldehydes to Nitroalkenes. Chem. Eur. J. 2009, 15, 9294–9298. [Google Scholar] [CrossRef]
- Chang, C.; Li, S.-H.; Reddy, R.J.; Chen, K. Pyrrolidine-Camphor Derivative as an Organocatalyst for Asymmetic Michael Additions of α,α-Disubstituted Aldehydes to β-Nitroalkenes: Construction of Quaternary Carbon-Bearing Aldehydes under Solvent-Free Conditions. Adv. Synth. Catal. 2009, 351, 1273–1278. [Google Scholar] [CrossRef]
- Ting, Y.-F.; Chang, C.; Reddy, R.J.; Magar, D.R.; Chen, K. Pyrrolidinyl–Camphor Derivatives as a New Class of Organocatalyst for Direct Asymmetric Michael Addition of Aldehydes and Ketones to β-Nitroalkenes. Chem. Eur. J. 2010, 16, 7030–7038. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Z.-H.; Chen, H.-Y.; Reddy, R.J.; Huang, C.-T.; Chen, K. Highly diastereo- and enantioselective direct aldol reactions promoted by water-compatible organocatalysts bearing a pyrrolidinyl-camphor structural scaffold. Tetrahedron 2009, 65, 2879–2888. [Google Scholar] [CrossRef]
- Bellis, E.; Kokotos, G. 4-Substituted prolines as organocatalysts for aldol reactions. Tetrahedron 2005, 61, 8669–8676. [Google Scholar] [CrossRef]
- Liu, P.-M.; Chang, C.; Reddy, R.J.; Ting, Y.-F.; Kuan, H.-H.; Chen, K. Remarkable Reaction Rate and Excellent Enantioselective Direct α-Amination of Aldehydes with Azodicarboxylates Catalyzed by Pyrrolidinylcamphor-Derived Organocatalysts. Eur. J. Org. Chem. 2010, 42–46. [Google Scholar] [CrossRef]
- Chen, Y.-m.; Lee, P.-H.; Lin, J.; Chen, K. Desymmetrization and Switching of Stereoselectivity in Direct Organocatalytic Michael Addition of Ketones to 1,1-Bis(phenylsulfonyl)ethylene. Eur. J. Org. Chem. 2013, 2013, 2699–2707. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Q.; Gong, Y. Camphor-derived C1-symmetric chiral diamine organocatalysts for asymmetric Michael addition of nitroalkanes to enones. Org. Biomol. Chem. 2012, 10, 7618–7627. [Google Scholar] [CrossRef]
- Lemay, M.; Ogilvie, W.W. Aqueous Enantioselective Organocatalytic Diels−Alder Reactions Employing Hydrazide Catalysts. A New Scaffold for Organic Acceleration. Org. Lett. 2005, 7, 4141–4144. [Google Scholar] [CrossRef]
- He, H.; Pei, B.-J.; Chou, H.-H.; Tian, T.; Chan, W.-H.; Lee, A.W.M. Camphor Sulfonyl Hydrazines (CaSH) as Organocatalysts in Enantioselective Diels−Alder Reactions. Org. Lett. 2008, 10, 2421–2424. [Google Scholar] [CrossRef]
- Li, Q.; Wong, W.-Y.; Chan, W.-H.; Lee, A.W.M. Second Generation CaSH (Camphor Sulfonyl Hydrazine) Organocatalysis. Asymmetric Diels–Alder Reactions and Isolation of the Catalytic Intermediate. Adv. Synth. Catal. 2010, 352, 2142–2146. [Google Scholar] [CrossRef]
- Li, Y.; Feng, Z.; You, S.-L. D-Camphor-derived triazolium salts for catalytic intramolecular crossed aldehyde–ketone benzoin reactions. Chem. Commun. 2008, 2263–2265. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.-Q.; Zheng, C.; You, S.-L. Highly enantioselective intramolecular Michael reactions by D-camphor-derived triazolium salts. Chem. Commun. 2009, 5823–5825. [Google Scholar] [CrossRef]
- Rong, Z.-Q.; Jia, M.-Q.; You, S.-L. Enantioselective N-Heterocyclic Carbene-Catalyzed Michael Addition to α,β-Unsaturated Aldehydes by Redox Oxidation. Org. Lett. 2011, 13, 4080–4083. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.-Q.; You, S.-L. N-Heterocyclic Carbene-Catalyzed Enantioselective Intramolecular N-Tethered Aldehyde–Ketone Benzoin Reactions. ACS Catal. 2013, 3, 622–624. [Google Scholar] [CrossRef]
- Jia, M.-Q.; You, S.-L. Desymmetrization of Cyclohexadienones via Intramolecular Stetter Reaction to Construct Tricyclic Carbocycles. Synlett 2013, 24, 1201–1204. [Google Scholar] [CrossRef]
- Rong, Z.-Q.; Li, Y.; Yang, G.-Q.; You, S.-L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett 2011, 1033–1037. [Google Scholar]
- Rafiński, Z.; Kozakiewicz, A. Enantioselective Synthesis of Chromanones Bearing Quaternary Substituted Stereocenters Catalyzed by (1R)-Camphor-Derived N-Heterocyclic Carbenes. J. Org. Chem. 2015, 80, 7468–7476. [Google Scholar] [CrossRef]
- Grošelj, U.; Ričko, S.; Svete, J.; Stanovnik, B. Synthesis of 2-(3-(3,5-Bis(trifluoromethyl)phenyl)thioureido)-3-((dimethylamino)methyl)camphor Organocatalysts. Chirality 2012, 24, 412–419. [Google Scholar] [CrossRef]
- Grošelj, U.; Sevšek, A.; Ričko, S.; Golobič, A.; Svete, J.; Stanovnik, B. Synthesis and Structural Characterization of Novel Camphor-derived Amines. Chirality 2012, 24, 778–788. [Google Scholar] [CrossRef]
- Ričko, S.; Golobič, A.; Svete, J.; Stanovnik, B.; Grošelj, U. Synthesis of Novel Camphor-Derived Bifunctional Thiourea Organocatalysts. Chirality 2015, 27, 39–52. [Google Scholar] [CrossRef]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, A.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.-Z.; Shao, Z.-H.; Fan, B.-M.; Song, H.; Li, G.-P.; Zhang, H.-B. Organocatalytic Enantioselective Michael Addition of 2,4-Pentanedione to Nitroalkenes Promoted by Bifunctional Thioureas with Central and Axial Chiral Elements. J. Org. Chem. 2008, 73, 5202–5205. [Google Scholar] [CrossRef]
- Almaşi, D.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Chiral 2-Aminobenzimidazoles as Recoverable Organocatalysts for the Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lee, M.-M.; Lee, S.-M.; Lee, J.; Cheng, M.; Jeong, B.-S.; Park, H.-g.; Jew, S.-s. Novel cinchona-aminobenzimidazole bifunctional organocatalyst. Adv. Synth. Catal. 2009, 351, 3063–3066. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Siva, A. Cinchona alkaloid-based chiral catalysts act as highly efficient multifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroolefins. Org. Biomol. Chem. 2015, 13, 10216–10225. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.Y.; Song, C.E. Unprecedented Hydrophobic Amplification in Noncovalent Organocatalysis “on Water”: Hydrophobic Chiral Squaramide Catalyzed Michael Addition of Malonates to Nitroalkenes. ACS Catal. 2015, 5, 3613–3619. [Google Scholar] [CrossRef]
- Jiménez, E.I.; Vallejo Narváez, W.E.; Román-Chavarría, C.A.; Vazquez-Chavez, J.; Rocha-Rinza, T.; Hernández-Rodríguez, M. Bifunctional Thioureas with α-Trifluoromethyl or Methyl Groups: Comparison of Catalytic Performance in Michael Additions. J. Org. Chem. 2016, 81, 7419–7431. [Google Scholar] [CrossRef]
- Veverková, E.; Bilka, S.; Baran, R.; Šebesta, R. Squaramide-Catalyzed Michael Addition as a Key Step for the Direct Synthesis of GABAergic Drugs. Synthesis 2016, 48, 1474–1482. [Google Scholar]
- Ñíguez, D.R.; Guillena, G.; Alonso, D.A. Chiral 2-Aminobenzimidazoles in Deep Eutectic Mixtures: Recyclable Organocatalysts for the Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to β-Nitroalkenes. ACS Sustainable Chem. Eng. 2017, 5, 10649–10656. [Google Scholar] [CrossRef] [Green Version]
- Tukhvatshin, R.S.; Kucherenko, A.S.; Nelyubina, Y.V.; Zlotin, S.G. Tertiary Amine-Derived Ionic Liquid-Supported Squaramide as a Recyclable Organocatalyst for Noncovalent “On Water” Catalysis. ACS Catal. 2017, 7, 2981–2989. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Knox, L.H. DL-Ketopinic acid. Org. Synth. 1965, 45, 55–56. [Google Scholar]
- Bartlett, P.D.; Knox, L.H. D,L-10-Camphorsulfonyl chloride. Org. Synth. 1965, 45, 14–16. [Google Scholar]
- Spino, C.; Joly, M.-A.; Godbout, C.; Arbour, M. Ti-Catalyzed Reactions of Hindered Isocyanates with Alcohols. J. Org. Chem. 2005, 70, 6118–6121. [Google Scholar] [CrossRef] [PubMed]
- Ishidate, M.; Kawada, A. Cleavage of the camphor ring. V. 7-Hydroxy-π-apocamphane and 1-hydroxy-10-apocamphor. Pharm. Bull. 1956, 4, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.-L.; Wu, H.-L.; Wu, P.-Y.; Uang, B.-J. Asymmetric addition of diethylzinc to aldehydes catalyzed by a camphor-derived β-amino alcohol. Tetrahedron Asymmetry 2009, 20, 1556–1560. [Google Scholar] [CrossRef]
- Grošelj, U.; Golobič, A.; Knez, D.; Hrast, M.; Gobec, S.; Ričko, S.; Svete, J. Synthesis and preliminary biological evaluations of (+)-isocampholenic acid-derived amides. Mol. Diversity 2016, 20, 667–676. [Google Scholar] [CrossRef]
- Garcia Martínez, A.; Teso Vilar, E.; Garcia Fraile, A.; de la Moya Cerero, S.; Diaz Oliva, C.; Subramanian, L.R.; Maichle, C. Synthesis of homochiral cyclopentane derivatives by Beckmann fragmentation of 1-substituted 2-norbornanones. Tetrahedron Asymmetry 1994, 5, 949–954. [Google Scholar] [CrossRef]
- Garcia Martínez, A.; Teso Vilar, E.; Garcia Fraile, A.; de la Moya Cerero, S.; Ruiz, P.M.; Subramanian, L.R. Synthesis of homochiral 3-substituted cyclopentanones from 2-norbornanones. Tetrahedron Asymmetry 1996, 7, 2177–2180. [Google Scholar] [CrossRef]
- Free amine could not be isolated in pure form. The crude amine obtained upon reduction of the respective oxime was subsequently transformed with 3,5-bis(trifluoromethyl)phenylisothiocyanate into isolable pure thiourea derivative.
- The crude compound obtained after given workup was used in the following transformation without further purification.
- García Martinez, A.; Teso Vilar, E.; García Fraile, A.; de la Moya Cerero, S.; Díaz Morillo, C. Enantiospecific access to 10-N-substituted camphors. Tetrahedron 2005, 61, 599–601. [Google Scholar] [CrossRef]
- García Martinez, A.; de la Moya Cerero, S.; Teso Vilar, E.; García Fraile, A.; Díaz Morillo, C.; de las Casas Engel, T. First efficient synthesis of enantiopure homoketopinic acid. Lett. Org. Chem. 2007, 4, 123–125. [Google Scholar] [CrossRef]
- Loudon, J.D. Mercury derivatives of camphor. I. The constitution of Reychler’s acid. J. Chem. Soc. 1933, 823–825. [Google Scholar] [CrossRef]
- Schenone, P.; Tasca, A.; Bignardi, G.; Mosti, L. N,N-disubstituted 10-amino-2-exo-bornanols and related esters. Eur. J. Med. Chem. Chim. Ther. 1975, 10, 412–417. [Google Scholar]
- Helliwell, M.; Thomas, E.J.; Townsend, L.A. Synthesis of chiral organotin reagents: Synthesis of enantiomerically enriched bicyclo[2.2.1]hept-2-yl tin hydrides from camphor. X-ray crystal structures of (dimethyl)[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl]tin chloride and methyl(phenyl)bis[(1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]hept-2-yl]stannane. J. Chem. Soc. Perkin Trans. 2002, 1286–1296. [Google Scholar]
- Grošelj, U.; Golobič, A.; Svete, J.; Ričko, S. Synthesis and reduction of 10-phthalimidocamphor oxime. Acta Chim. Slov. 2017, 64, 790–797. [Google Scholar] [CrossRef] [Green Version]
- For details see the Supporting Information.
- Page, P.C.B.; Murrell, V.L.; Limousin, C.; Laffan, D.D.P.; Bethell, D.; Slawin, A.M.Z.; Smith, T.A.D. The first stable enantiomerically pure chiral N-H oxaziridines: Synthesis and reactivity. J. Org. Chem. 2000, 65, 4204–4207. [Google Scholar] [CrossRef]
- Ju, Y.; Varma, R.S. An Efficient and Simple Aqueous N-Heterocyclization of Aniline Derivatives: Microwave-Assisted Synthesis of N-Aryl Azacycloalkanes. Org. Lett. 2005, 7, 2409–2411. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio- and Diastereoselective Michael Reaction of 1,3-Dicarbonyl Compounds to Nitroolefins Catalyzed by a Bifunctional Thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef]
- Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. Theoretical Studies on the Bifunctionality of Chiral Thiourea-Based Organocatalysts: Competing Routes to C−C Bond Formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef]
- Tan, B.; Lu, Y.; Zeng, X.; Chua, P.J.; Zhong, G. Facile Domino Access to Chiral Bicyclo[3.2.1]octanes and Discovery of a New Catalytic Activation Mode. Org. Lett. 2010, 12, 2682–2685. [Google Scholar] [CrossRef]
- Sohtome, Y.; Shin, B.; Horitsugi, N.; Takagi, R.; Noguchi, K.; Nagasawa, K. Entropy-Controlled Catalytic Asymmetric 1,4-Type Friedel–Crafts Reaction of Phenols Using Conformationally Flexible Guanidine/Bisthiourea Organocatalyst. Angew. Chem. Int. Ed. 2010, 49, 7299–7303. [Google Scholar] [CrossRef] [PubMed]
- Sohtome, Y.; Horitsugi, N.; Takagi, R.; Nagasawa, K. Enantioselective Phospha-Michael Reaction of Diphenyl Phosphonate with Nitroolefins Utilizing Conformationally Flexible Guanidinium/Bisthiourea Organocatalyst: Assembly-State Tunability in Asymmetric Organocatalysis. Adv. Synth. Catal. 2011, 353, 2631–2636. [Google Scholar] [CrossRef]
- Yao, W.; Chen, M.; Liu, X.; Jiang, R.; Zhang, S.; Chen, W. Ferrocene as a scaffold for effective bifunctional amine–thiourea organocatalysts. Catal. Sci. Technol. 2014, 4, 1726–1729. [Google Scholar] [CrossRef]
- Ren, X.; He, C.; Feng, Y.; Chai, Y.; Yao, W.; Chen, W.; Zhang, S. Novel ferrocene-based bifunctional amine–thioureas for asymmetric Michael addition of acetylacetone to nitroolefins. Org. Biomol. Chem. 2015, 13, 5054–5060. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Abe, T.; Wang, X.-B.; Kodama, K.; Hirose, T.; Zhang, G.-Y. Self-assembled proline-amino thioureas as efficient organocatalysts for the asymmetric Michael addition of aldehydes to nitroolefins. Tetrahedron Asymmetry 2010, 21, 2925–2933. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 18, 20a–23a, 24, 52, and 53 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Temperature (°C) | Conversion (%) | Er |
| 1 | 48 | 25 | 38 | 68.5:31.5; (S) |
| 2 | 48 | −25 | 30 | 66.5:33.5; (S) |
| 3 | 49 | 25 | 61 | 61:39; (R) |
| 4 | 49 | −25 | 16 | 53:47; (R) |
| 5 | 50 | 25 | 20 | 66.5:33.5; (R) |
| 6 | 50 | −25 | 6 | 61:39; (R) |
| 7 | 51 | 25 | 5 | 57:43; (S) |
| 8 | 51 | −25 | 2 | 59:41; (S) |
| 9 | 52 | 25 | >99 | 78.5:21.5; (R) |
| 10 | 52 | −25 | 90 | 78.5:21.5; (R) |
| 11 | 53 | 25 | 69 | 77.5:22.5; (S) |
| 12 | 53 | −25 | 15 | 66.5:33.5; (S) |
| 13 | 54 | 25 | 80 | 61.5:38:5; (R) |
| 14 | 54 | −25 | 64 | 55:45; (R) |
| 15 | 55 | 25 | 10 | 54.5:45.5; (R) |
| 16 | 56 | 25 | >99 | 63:37; (R) |
| 17 | 56 | −25 | >99 | 61.5:38.5; (R) |
| 18 | 57 | 25 | 79 | 50.5:49.5 (R) |
| 19 | 58 | 25 | 85 | 50.5:49.5 (S) |
| 20 2 | 59 | 25 | 99 | 66.5:33.5; (S) |
| 21 | 59 | −25 | 61 | 64:36; (S) |
| 22 2 | 60 | 25 | 9 | 56:44; (S) |
| 23 | 60 | −25 | 6 | 67:33; (S) |
| 24 | 61 | −25 | 93 | 55:45; (R) |
| 25 | 62 | 25 | >99 | 57.5:42.5; (S) |
| 26 | 62 | −25 | 74 | 62.5:37.5; (S) |
| 27 | 63 | 25 | >99 | 54.5:45.5; (S) |
| 28 | 63 | −25 | >99 | 50.5:49.5; (S) |
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | R | Temperature (°C) | Conversion (%) | Er |
| 1 | 48 | Me | 25 | 96 | 69:31; (S) |
| 2 | 49 | Me | 25 | >99 | 80.5:19.5; (R) |
| 3 | 52 | Me | 25 | >99 | 87:13; (S) |
| 4 | 52 | Me | −25 | >99 | 91.5:8.5; (S) |
| 5 2 | 52 | Me | 25 | >99 | 74.5:25.5; (S) |
| 6 3 | 52 | Me | 25 | >99 | 81:19; (S) |
| 7 | 52 | Ph | 25 | >99 | 87:13; (S) |
| 8 | 53 | Me | 25 | 96 | 79.5:20.5; (R) |
| 9 | 53 | Me | −25 | 63 | 82:18; (R) |
| 10 | 53 | Ph | 25 | >99 | 81:19; (R) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ričko, S.; Požgan, F.; Štefane, B.; Svete, J.; Golobič, A.; Grošelj, U. Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules 2020, 25, 2978. https://doi.org/10.3390/molecules25132978
Ričko S, Požgan F, Štefane B, Svete J, Golobič A, Grošelj U. Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules. 2020; 25(13):2978. https://doi.org/10.3390/molecules25132978
Chicago/Turabian StyleRičko, Sebastijan, Franc Požgan, Bogdan Štefane, Jurij Svete, Amalija Golobič, and Uroš Grošelj. 2020. "Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts" Molecules 25, no. 13: 2978. https://doi.org/10.3390/molecules25132978
APA StyleRičko, S., Požgan, F., Štefane, B., Svete, J., Golobič, A., & Grošelj, U. (2020). Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules, 25(13), 2978. https://doi.org/10.3390/molecules25132978