3.2. Synthesis
2-(3-((4-Methoxyphenyl)amino)propanoyl)-N-phenylhydrazinecarboxamide (2): A mixture of propane-hydrazide (1, 2.09 g, 10 mmol), methanol (30 mL), and phenyl isocyanate (1.35 g, 1.08 mL, 10 mmol) was heated at reflux for 30 min. The reaction mixture was cooled down, precipitate formed was filtered off and recrystallized from DMF/H2O mixture. Yield 59%; white solid; m.p. 192–193 °C; IR (KBr) νmax (cm−1): 3375, 3344, 3283, 3206 (NH), 1659, 1638 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.41 (t, J = 7.2 Hz, 2H, CH2CO), 3.78 (s, 3H, CH3O), 3.84 (t, J = 7.2 Hz, 2H, CH2NH), 6.99–7.44 (m, 9H, HAr), 7.76, 8.01, 8.71, 9.77 (4s, 4H, 4NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 32.3 (CH2CO), 46.4 (CH2NH), 55.3 (CH3O), 114.9, 119.9, 128.3, 128.7, 129.7, 134.1, 139.6, 140.0, 154.7 (CAr), 158.1, 170.3 (C=O); HRMS (ESI): m/z calcd for C17H20N4O3 328.1535 [M + H]+, found 328.1537.
2-(3-((4-Methoxyphenyl)amino)propanoyl)-N-phenylhydrazinecarbothioamide (3): A mixture of propanehydrazide 1 (3.15 g, 15 mmol), methanol (60 mL), and phenyl isothiocyanate (0.42 g, 3.71 mL, 20 mmol) was heated at reflux for 4 h. The reaction mixture was cooled down, precipitate formed was filtered off and recrystallized from DMF/H2O mixture. Yield 78%; white solid; m.p. 164–165 °C; IR (KBr) νmax (cm−1): 3451–2831 (NH), 1698 (C=O), 1244 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.46 (t, J = 7.2 Hz, 2H, CH2CO), 3.24 (t, J = 7.2 Hz, 2H, CH2NH); 3.63 (s, 3H, CH3O), 5.18 (s, 1H, NH), 6.55 (d, J = 8.8 Hz, 2H, HAr2,6), 6.73 (d, J = 8.8 Hz, 2H, HAr3,5), 7.21–7.37 (m, 5H, HAr‘), 9.54, 9.59 (2s, 2H, NHNHCS), 9.98 (s, 1H, NHAr‘); 13C-NMR (DMSO-d6, 101 MHz) δ: 33.5 (CH2CO), 40.2 (CH2NH), 55.3 (CH3O), 113.4, 114.7, 115.29, 126.1, 127.9, 139.1, 142.9, 150.9 (CAr), 158.6 (C=O), 180.8 (C=S); HRMS (ESI): m/z calcd for C17H20N4O2S 345.1385 [M + H]+, found 345.1379.
3.2.1. General Procedure for Synthesis of Compounds 4 and 5
To propanehydrazide 1 (2.09 g, 0.01 mol) dissolved in methanol (70 mL), corresponding cyanate (0.02 mol) was added. The reaction mixture was heated at reflux for 1–2 h. Precipitate formed was filtered off, washed with water, and recrystallized from DMF/H2O mixture.
2-(3-(1-(4-Methoxyphenyl)-3-phenylureido)propanoyl)-N-phenylhydrazinecarboxamide (4): Prepared from phenyl isocyanate by heating at reflux a reaction mixture for 2 h. Yield 76%; white solid; m.p. 179–180 °C; IR (KBr) νmax (cm−1): 3386, 3254, 3283, 3206 (NH); 1675, 1661, 1601 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.43 (t, J = 7.2 Hz, 2H, CH2CO), 3.79 (s, 3H, CH3O), 3.85 (t, J = 7.2 Hz, 2H, CH2N), 6.88–7.48 (m, 14H, HAr, Ar’, Ar’’), 7.72 (s, 1H, NH), 8.00 (s, 1H, NH), 8.69 (s, 1H, NH), 9.76 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 32.3 (CH2CO), 46.2 (CH2N), 55.3 (CH3O), 114.9, 118.6, 119.8, 121.9, 122.0, 128.2, 128.6, 129.4, 129.6, 134.1, 139.6, 139.9, 154.6 (CAr), 155.3, 158.1, 170.5 (C=O); HRMS (ESI): m/z calcd for C24H25N5O4 447.1985 [M + H]+, found 448.1987.
2-(3-(1-(4-Methoxyphenyl)-3-phenylthioureido)propanoyl)-N-phenylhydrazinecarbothioamide (5): Prepared from phenyl isothiocyanate by heating at reflux a reaction mixture for 1 h. Yield 78%; white solid; m.p. 178–179 °C; IR (KBr) νmax (cm−1): 3333, 3299, 3206, 3154 (NH), 1698 (C=O), 1243, 1216 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.64 (t, J = 7.8 Hz, 2H, CH2CO), 3.77, 3.78 (2s, 3H, CH3O), 4.34 (t, J = 7.8 Hz, 2H, CH2N), 6.96–7.34 (m, 14H, HAr, Ar’, Ar’’), 7.38, 8.65, 9.50, 9.95 (4s, 4H, 4NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 31.0 (CH2CO), 50.6 (CH2N), 55.4 (CH3O), 115.1, 115.2, 124.7, 125.8, 126.0, 127.8, 128.0, 129.1, 134.7, 139.1, 140.5, 158.6 (CAr), 170.20 (C=O), 180.8, 181.7 (C=S); HRMS (ESI): m/z calcd for C24H25N5O2S2 480.1528 [M + H]+, found 480.1526.
1-(4-Methoxyphenyl)-3-phenyl-1-(2-(5-(phenylamino)-1,3,4-thiadiazol-2-yl)ethyl)thiourea (6): A mixture of conc. H2SO4 (25 mL) and phenylhydrazinecarbothioamide 5 (0.96 g, 2 mmol) was stirred at room temperature until all solid dissolved (approx. 25 min). Afterwards the reaction mixture was added drop by drop to a water-ice mixture. Precipitate formed was filtered off, washed with water, and recrystallized from propan-2-ol. Yield 77%; white solid; m.p. 151–152 °C; IR (KBr) νmax (cm−1): 3358, 3191 (NH), 1249 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 3.29 (t, J = 7.4 Hz, 2H, CH2C), 3.76 (s, 3H, CH3O), 4.46 (t, J = 7.4 Hz, 2H, CH2N), 6.96–7.59 (m, 14H, HAr, Ar’, Ar’’), 8.66 (s, 1H, NH), 10.29 (s, 1H, NHC=S); 13C-NMR (DMSO-d6, 101 MHz) δ: 27.8 (CH2C), 53.8 (CH2N), 55.3, 55.5 (CH3O), 115.0, 115.3, 117.2, 121.7, 124.9, 126.3, 127.8, 129.1, 134.6, 140.5, 140.8, 156.4, 158.6, 164.4 (CAr), 181.9 (C=S); HRMS (ESI): m/z calcd for C24H23N5OS2 462.1422 [M + H]+, found 462.1419.
3-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-1H-1,2,4-triazol-5(4H)-one (7): A mixture of phenyl-hydrazinecarboxamide 2 (1.34 g, 3 mmol) and 20% aqueous KOH solution (25 mL) was heated at reflux for 2 h. The reaction mixture was cooled down and acidified with HCl to pH 4. Precipitate formed was filtered off, washed with water, and recrystallized from DMF/H2O mixture. Yield 81%; white solid; m.p. 156–157 °C; IR (KBr) νmax (cm−1): 3175, 3017 (NH), 1659 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.62 (t, J =7.2 Hz, 2H, CH2C), 3.14 (t, J =7.2 Hz, 2H, CH2NH), 3.62 (s, 3H, CH3O); 6.39 (d, J = 8.4 Hz, 2H, HAr2,6), 6.67 (d, J = 8.4 Hz, 2H, HAr3,5), 7.41–7.57 (m, 6H, HAr’+NH), 11.76 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 25.6 (CH2C), 40.8 (CH2NH), 55.3 (CH3O), 113.2, 114.7, 127.6, 128.8, 129.6, 133.0, 145.2, 145.3, 150.9, 151.5 (CAr), 154.4 (C=O); HRMS (ESI): m/z calcd for C17H18N4O2 311.1509 [M + H]+, found 311.1503.
3-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-1H-1,2,4-triazole-5(4H)-thione (8): A mixture of phenyl-hydrazinecarbothioamide 3 (1 g, 3 mmol) and 20% aqueous KOH solution (40 mL) was heated at reflux for 4 h. The reaction mixture was cooled down and acidified with HCl to pH 4. Precipitate formed was filtered off, washed with water, and recrystallized from DMF/H2O mixture. Yield 80%; white solid; m.p. 141–142 °C; IR (KBr) νmax (cm−1): 3446–2826 (NH), 1237 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.63 (t, J = 9.0 Hz, 2H, CH2C), 3.15 (t, 2H, J = 9.0 Hz, CH2NH), 3.61 (s, 3H, CH3O), 5.22 (s, 1H, NHAr’), 6.31 (d, J = 9 Hz, 2H, HAr2,6), 6.63 (d, J = 9 Hz, 2H, HAr3,5), 7.39–7.58 (m, 5H, HAr’), 13.75 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 25.4 (CH2C), 40.4 (CH2NH), 55.3 (CH3O), 113.1, 114.7, 122.0, 128.5, 129.5, 133.8, 142.2, 150.7, 150.9, 167.6 (CAr), 172.1 (C=S); HRMS (ESI): m/z calcd for C17H18N4OS 327.1279 [M + H]+, found 327.1293.
3.2.2. General Procedure for the Synthesis of 1,2,4-Triazolones 9 and 10
To hydrazinecarboxamide 7 (0.93 g, 3 mmol) dissolved in DMF (15 mL), KOH (0.17 g, 3 mmol), K2CO3 (0.37 g, 2.7 mmol), and corresponding bromoacetophenone (3.75 mmol) were added. The reaction mixture was stirred at 40 °C for 24 h. Afterwards water (30 mL) was added to the reaction mixture. Precipitate formed was filtered off, washed with water, and recrystallized from propan-2-ol.
3-(2-((4-Methoxyphenyl)(2-oxo-2-phenylethyl)amino)ethyl)-4-phenyl-1H-1,2,4-triazol-5(4H)-one (9): Prepared from 2-bromoacetophenone. Yield 63%; white solid; m.p. 142–143 °C; IR (KBr) νmax (cm−1): 3375 (NH), 1694, 1513 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.70 (t, J = 7.4 Hz, 2H, CH2C), 3.49 (t, J = 7.4 Hz, 2H, CH2N), 3.61 (s, 3H, CH3O), 4.80 (s, 2H, CH2CO), 6.22 (d, J = 9.2 Hz, 2H, HAr2,6), 6.62 (d, J = 9.2 Hz, 2H, HAr3,5), 7.41–7.96 (m, 10H, HAr’, Ar’’), 11.68, 11.71 (2s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.1 (CH2C), 40.2 (CH2N), 48.4 (CH2CO), 55.3 (CH3O), 112.6, 114.6, 127.5, 127.6, 127.7, 128.7, 128.8, 129.5, 132.9, 133.4, 135.3, 141.6, 145.4, 150.7 (CAr), 154.3, 197.2 (C=O); HRMS (ESI): m/z calcd for C25H24N4O3 429.1927 [M + H]+, found 429.1930.
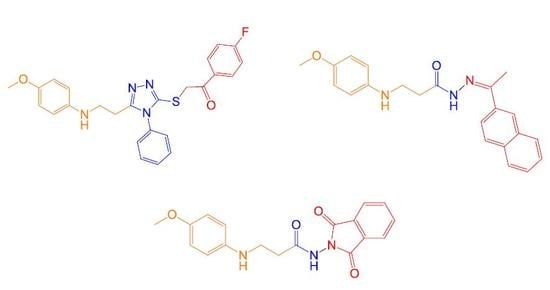
3-(2-((2-(4-Fluorophenyl)-2-oxoethyl)(4-methoxyphenyl)amino)ethyl)-4-phenyl-1H-1,2,4-triazol-5(4H)-one (10): Prepared from 2-bromo-4′-fluoroacetophenone. Yield 58%; white solid; m.p. 103–104 °C; IR (KBr) νmax (cm−1): 3494 (NH), 1698, 1514 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.70 (t, J = 7.4 Hz, 2H, CH2C), 3.48 (t, J = 7.4 Hz, 2H, CH2N), 3.61 (s, 3H, CH3O), 4.80 (s, 2H, CH2CO), 6.23 (d, J = 9.2 Hz, 2H, HAr2,6), 6.62 (d, J = 9.2 Hz, 2H, HAr3,5), 7.35–7.55 (m, 7H, HAr’, Ar’’), 8.02–8.05 (m, 2H, HAr’’), 11.67, 11.71 (2s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.1 (CH2C), 40.2 (CH2N), 48.3 (CH2CO), 55.3 (CH3O), 112.7, 114.6, 115.6, 115.9, 127.6, 128.7, 129.5, 130.7, 130.8, 132.0, 132.0, 132.9, 141.6, 145.3, 150.7, 154.3, 163.9 (CAr), 166.4, 195.8 (C=O); HRMS (ESI): m/z calcd for C25H23FN4O3 447.1833 [M + H]+, found 447.1830.
N-(4-Methoxyphenyl)-N-(2-(4-phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)ethyl)acetamide (11): A mixture of triazolethione 8 (2.61 g, 8 mmol) and acetyl chloride (25 mL) was heated at reflux for 7 h. The reaction mixture was added drop by drop onto ice. Precipitate formed was filtered off, washed with water, and recrystallized from acetone. Yield 66%; white solid; m.p. 182–183 °C; IR (KBr) νmax (cm−1): 1651, 1510 (C=O), 1255 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.62, 1.65 (2s, 3H, CH3), 2.60 (t, J = 6.8, 2H, CH2C), 3.56–3.70 (m, 2H, CH2N), 3.77 (s, 3H, CH3O), 6.94–7.56 (m, 9H, HAr); 13.70 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 22.2, 23.8 (CH3), 24.7 (CH2C), 45.4 (CH2N), 55.4 (CH3O), 114.7, 128.3, 129.2, 129.3, 129.5, 133.6, 135.0, 149.8, 158.4, 158.5 (CAr), 167.7 (C=O), 169.4 (C=S); HRMS (ESI): m/z calcd for C19H20N4O2S 369.1386 [M + H]+, found 369.1381.
3.2.3. General Procedure for the Synthesis of Compounds 12a, b
A mixture of triazolethione 7 or 8 (2 mmol), potassium thiocyanate (0.39 g, 4 mmol), and acetic acid (10 mL) was heated at reflux for 5 min. The reaction mixture was cooled to room temperature and diluted with water (50 mL). Precipitate formed was filtered off, washed with water, and recrystallized from propan-2-ol.
1-(4-Methoxyphenyl)-1-(2-(5-oxo-4-phenyl-4,5-dihydro-1H-1,2,4-triazol-3-yl)ethyl)thiourea (12a): Prepared from 7 (0.62 g). Yield 76%; white solid; m.p. 203–204 °C; IR (KBr) νmax (cm−1): 3242, 3176 (NH, NH2), 1691 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.73 (t, 2H, J = 7.2 Hz, CH2C), 3.34, 3.61 (2s, 2H, NH2), 3.77 (s, 3H, CH3O), 4.07 (t, J = 7.2 Hz, 2H, CH2N), 6.95 (d, J = 8.8 Hz, 2H, HAr2,6), 7.02 (d, J = 8.8 Hz, 2H, HAr3,5), 7.23–7.46 (m, 5H, HAr’), 11.67, 11.71 (2s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.2 (CH2C), 51.1 (CH2N), 55.4 (CH3O), 115.2, 127.2, 128.5, 128.8, 129.3, 132.7, 133.9, 144.4, 154.2 (CAr), 158.6 (C=O), 181.9 (C=S); HRMS (ESI): m/z calcd for C18H19N5O2S 370.1337 [M + H]+, found 370.1337.
1-(4-Methoxyphenyl)-1-(2-(4-phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)ethyl)thiourea (12b): Prepared from 8 (0.65 g). Yield 66%; white solid; m.p. 198–199 °C; IR (KBr) νmax (cm−1): 3233, 3185 (NH, NH2), 1520 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.73 (t, J = 7.2 Hz, 2H, CH2C), 3.34 (s, 2H, NH2), 3.77 (s, 3H, CH3O), 4.09 (t, J = 7.2 Hz, 2H, CH2N), 6.95 (d, J = 8.8 Hz, 2H, HAr2,6), 7.02 (d, 2H, J = 8.8 Hz, HAr3,5), 7.30–7.50 (m, 5H, HAr‘), 13.70 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 23.7 (CH2C), 51.2 (CH2N), 55.4 (CH3O), 114.7, 115.2, 128.1, 128.8, 129.3, 129.4, 133.5, 133.7, 149.6, 158.6 (CAr), 167.6, 181.9 (C=S); HRMS (ESI): m/z calcd for C18H19N5OS2 386.1109 [M + H]+, found 386.1104.
2-(2-((4-Methoxyphenyl)(2-(4-phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)ethyl)amino)-4-oxo-4,5-dihydrothiazol-5-yl)acetic acid (13): A mixture of 12b (0.58 g, 1.5 mmol), maleic anhydride (0.29 g, 3 mmol), 1,4-dioxane (15 mL), and DMF (5 mL) was heated at reflux for 7 h. The reaction mixture was cooled down and cold water (10 mL) was added. Precipitate formed was filtered off and recrystallized from DMF/H2O mixture. Yield 82%; yellow solid; m.p. 159–160 °C; IR (KBr) νmax (cm−1): 3233, 3185 (NH, NH2), 1648, 1713 (C=O), 1530 (C=S); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.52–2.60 (m, 1H, CH), 2.73–2.83 (m, 2H, CH2C), 2.98–3.05 (m, 1H, CH2), 3.81 (s, 3H, CH3O), 3.88–4.09 (m, 2H, CH2N), 4.23–4.35 (m, 1H, CH2), 6.98–7.57 (m, 9H, HAr, Ar‘), 12.78 (br s, 1H, OH), 13.75 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 23.6 (CH2C), 37.6 (CH2), 50.3 (CH2N), 52.2 (CS), 55.6 (CH3O), 114.9, 128.3, 129.3, 129.4, 129.5, 132.5, 133.4, 149.1, 159.7 (CAr), 167.8 (C=S), 172.4 (COOH), 183.0 (NCS), 187.4 (C=O); HRMS (ESI): m/z calcd for C22H21N5O4S2 484.1113 [M + H]+, found 484.1110.
3.2.4. General Procedure for the Synthesis of S-Substituted 1,2,4-Triazolethiones 14–24
Method A. To triazolethione 8 (0.49 g, 1.5 mmol) dissolved in DMF (5 mL), triethylamine (0.20 g, 0.28 mL, 2 mmol) and corresponding halocarbonyl compound (2 mmol) were added. The reaction mixture was stirred at room temperature for 4 h. Afterwards cold water (30 mL) was added, the precipitate formed was filtered off and recrystallized from propan-2-ol.
Method B. To triazolethione 8 (0.49 g, 1.5 mmol) dissolved in DMF (5 mL), KOH powder (0.11 g, 2 mmol), K2CO3 (0.28 g, 2.2 mmol), and corresponding halocarbonyl compound (2 mmol) were added. The reaction mixture was stirred at 35–40 °C for 24 h. Afterwards cold water (30 mL) was added, the precipitate formed was filtered off and recrystallized from propan-2-ol.
Method C. To triazolethione 8 (0.49 g, 1.5 mmol) dissolved in acetone (15 mL), K2CO3 (1 g, 7.2 mmol) and corresponding halocarbonyl compound (2 mmol) were added. The reaction mixture was stirred at 40 °C for 3 h. Afterwards water (20 mL) was added, the precipitate formed was isolated by filtration, and recrystallized from propan-2-ol.
((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl propionate (14): Prepared according to method A from ethyl chloroacetate (0.25 g, 0.21 mL). Yield 80%; white solid; m.p. 71–72 °C; IR (KBr) νmax (cm−1): 3309 (NH), 1753 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.17 (t, J = 7.2 Hz, 3H, CH3CH2), 2.73 (t, J = 7.2 Hz, 2H, CH2C), 3.19 (t, 2H, J = 7.2 Hz, 2H, CH2NH), 3.61, 3.62 (2s, 3H, CH3O), 4.01 (s, 2H, SCH2), 4.09 (q, J = 7.2 Hz, 2H, CH3CH2), 5.24 (s, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.44–7.64 (m, 5H, HAr’); 13C-NMR (DMSO-d6, 101 MHz) δ: 14.0 (CH3CH2), 24.7 (CH2C), 33.9 (SCH2), 41.3 (CH2NH); 55.3 (CH3O), 61.2 (CH3CH2), 112.6, 113.1, 114.6, 127.4, 130.0, 130.1, 132.8, 142.2, 149.1, 150.8 (CAr), 154.2 (C=O), 168.1 (C-S-); HRMS (ESI): m/z calcd for C21H24N4O3S 413.1647 [M + H]+, found 413.1660.
2-((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (15): Prepared according to method B from 2-chloroacetamide (0.19 g). Yield 75%; white solid; m.p. 77–78 °C; IR (KBr) νmax (cm−1): 3346–2826 (NH, NH2), 1696 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.2 Hz, 2H, CH2C), 3.20 (q, 2H, J = 7.2 Hz, CH2NH), 3.62 (s, 3H, CH3O), 3.88 (s, 2H, SCH2), 5.23 (t, J = 6.2 Hz, 1H, NH), 6.32 (d, J = 8.8 Hz, 2H, HAr2,6), 6.64 (d, J = 8,8 Hz, 2H, HAr3,5), 7.23 (s, 1H, NH2), 7.46–7.61 (m, 5H, HAr’), 7.67 (s, 1H, NH2); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.7 (CH2C), 35.9 (SCH2), 41.3 (CH2NH), 55.3 (CH3O), 113.1, 114.6, 127.4, 129.9, 130.1, 132.9, 142.2, 149.9, 150.8 (CAr), 154.0 (C=O), 168.7 (C-S-); HRMS (ESI): m/z calcd for C19H21N5O2S 384.1494 [M + H]+, found 384.1500.
2-((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-1-phenylethanone (16): Prepared according to method A from 2-bromoacetophenone (0.4 g). Yield 69%; white solid; m.p. 159–160 °C; IR (KBr) νmax (cm−1): 3292 (NH), 1687 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.2 Hz, 2H, CH2C), 3.15–3.25 (m, 2H, CH2NH), 3.61 (s, 3H, CH3O), 4.86 (s, 2H, SCH2), 5.23 (s, 1H, NH), 6.32 (d, J = 8.8 Hz, 2H, HAr2,6), 6.64 (d, J = 8.8 Hz, 2H, HAr3,5), 7.46–8.02 (m, 10H, HAr’, Ar”); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.6 (CH2C), 40.0 (SCH2), 41.2 (CH2NH), 55.2 (CH3O), 112.9, 114.5, 127.3, 128.3, 128.7, 129.9, 132.8, 133.6, 135.2, 142.1, 149.3, 150.7 (CAr), 154.0 (C=O), 193.1 (C-S-); HRMS (ESI): m/z calcd for C25H24N4O2S 445.1698 [M + H]+, found 445.1708.
1-(4-Bromophenyl)-2-((5-(2-((4-methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethanone (17): Prepared according to method A from 4-bromophenacyl bromide (0.56 g). Yield 63%; white solid; m.p. 168–169 °C; IR (KBr) νmax (cm−1): 3291 (NH), 1694 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.73 (t, J = 7.2 Hz, 2H, CH2C), 3.19 (t, 2H, J = 7.2 Hz, CH2NH), 3.61 (s, 3H, CH3O), 4.82 (s, 2H, SCH2), 5.22 (s, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.45–7.94 (m, 9H, HAr’, Ar”); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.8 (CH2C), 40.2 (SCH2), 41.3 (CH2NH), 55.3 (CH3O), 113.1, 114.6, 127.4, 127.8, 130.0, 130.1, 130.4, 131.9, 132.9, 134.3, 142.2, 149.2, 150.8 (CAr), 154.2 (C=O), 192.5 (C-S-); HRMS (ESI): m/z calcd for C25H23BrN4O2S 523.0803 [M + H]+, found 523.0799.
2-((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-1-(p-tolyl)ethanone (18): Prepared according to method C from 2-bromo-4′-methylacetophenone (0.43 g). Yield 71%; white solid; m.p. 136–137 °C; IR (KBr) νmax (cm−1): 3282 (NH), 1689 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.39 (s, 3H, CH3), 2.74 (t, J = 7.4 Hz, 2H, CH2C), 3.19 (q, J = 6.8, 14.0 Hz, 2H, CH2NH), 3.61 (s, 3H, CH3O), 4.82 (s, 2H, SCH2), 5.22 (t, J = 6.0 Hz, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.35 (d, J = 8.0 Hz, 2H, HAr’’), 7.45–7.50 (m, 2H, HAr’), 7.58–7.63 (m, 3H, HAr’), 7.90 (d, J = 8.0 Hz, 2H, HAr’’); 13C-NMR (DMSO-d6, 101 MHz) δ: 21.2 (CH3), 24.7 (CH2C), 40.1 (SCH2), 41.3 (CH2NH), 55.3 (CH3O), 113.1, 114.6, 127.4, 128.5, 129.4, 130.0, 130.1, 132.8, 132.9, 142.2, 144.3, 149.4, 150.8 (CAr), 154.1 (C=O), 192.7 (C-S-); HRMS (ESI): m/z calcd for C26H26N4O2S 459.1854 [M + H]+, found 459.1853.
1-(4-Methoxyphenyl)-2-((5-(2-((4-methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethanone (19): Prepared according to method C from 2-bromo-4’-methoxyacetophenone (0.30 g). Yield 76%; white solid; m.p. 121–122 °C; IR (KBr) νmax (cm−1): 3287 (NH), 1683 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.76 (t, J = 7.2 Hz, 2H, CH2C), 3.25 (t, J = 7.2 Hz, 2H, CH2NH), 3.61, 3.64 (2s, 3H, CH3O), 3.85, 3.86 (2s, 3H, CH3O), 4.79, 4.80 (2s, 2H, SCH2), 5.22 (s, 1H, NH), 6.45–7.99 (m, 14H, HAr, Ar‘, Ar‘‘+NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.3 (CH2C), 40.0 (SCH2), 42.2 (CH2NH), 55.3, 55.3 (CH3O), 55.6, 55.6 (CH3O), 112.5, 114.0, 114.7, 127.4, 128.1, 130.0, 130.1, 130.8, 132.9, 141.6, 149.6, 150.6, 153.8 (CAr), 163.6 (C=O), 191.5 (C-S-); HRMS (ESI): m/z calcd for C26H26N4O3S 475.1804 [M + H]+, found 475.1802.
1-(4-Chlorophenyl)-2-((5-(2-((4-methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethanone (20): Prepared according to method A from 2-bromo-4’-chloroacetophenone (0.47 g). Yield 61%; white solid; m.p. 152–153 °C; IR (KBr) νmax (cm−1): 3291 (NH), 1694 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.73 (t, J = 7.2 Hz, 2H, CH2C), 3.19 (t, 2H, J = 7.2 Hz, CH2NH), 3.61 (s, 3H, CH3O), 4.83 (s, 2H, SCH2), 5.25 (s, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.44–8.06 (m, 9H, HAr′, Ar″); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.72 (CH2C), 40.22 (SCH2), 41.29 (CH2NH), 55.28 (CH3O), 113.10, 114.62, 127.37, 128.92, 129.80, 130.00, 130.07, 130.32, 132.90, 134.01, 138.62, 142.15, 149.24, 150.82 (CAr), 154.15 (C=O), 192.29 (C-S-); HRMS (ESI): m/z calcd for C25H23ClN4O2S 479.1308 [M + H]+, found 479.1306.
1-(4-Fluorophenyl)-2-((5-(2-((4-methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethanone (21): Prepared according to method C from 2-bromo-4′-fluoroacetophenone (0.43 g). Yield 73%; white solid; m.p. 145–146 °C; IR (KBr) νmax (cm−1): 3290 (NH); 1693 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.4 Hz, 2H, CH2C), 3.19 (t, J = 7.4 Hz, 2H, CH2NH), 3.61 (s, 3H, CH3O), 4.84 (s, 2H, SCH2), 5.22 (s, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.36–7.61 (m, 7H, HAr’, Ar’’), 8.07–8.11 (m, 2H, HAr’’); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.7 (CH2C), 40.2 (SCH2), 41.3 (CH2NH), 55.3 (CH3O), 113.1, 114.6, 115.8, 115.9, 127.4, 130.0, 130.1, 131.4, 132.1, 132.9, 142.2, 149.3, 150.8 (CAr), 154.1 (C=O), 191.8 (C-S-); HRMS (ESI): m/z calcd for C25H23FN4O2S 463.1604 [M + H]+, found 463.1601.
2-((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-1-(4-nitrophenyl)ethanone (22): Prepared according to method C from 2-bromo-4′-nitroacetophenone (0.49 g), recrystallized from propan-2-ol. Yield 88 %; brown solid; m.p. 169–170 °C IR (KBr) νmax (cm−1): 3306 (NH), 1698 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.4 Hz, 2H, CH2C), 3.19 (t, J = 7.0 Hz, 2H, CH2NH), 3.61 (s, 3H, CH3O), 4.89 (s, 2H, SCH2), 5.22 (s, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.46–7.49 (m, 2H, HAr’), 7.60–7.62 (m, 3H, HAr’), 8.23 (d, J = 8,7 Hz, 2H, HAr’’), 8.36 (d, J = 8.7 Hz, 2H, HAr’’); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.7 (CH2C), 40.2 (CH2NH), 41.3 (SCH2), 55.3 (CH3O), 113.1, 114.6, 123.9, 127.4, 129.8, 130.0, 130.1, 132.9, 140.1, 142.2, 149.1, 150.1, 150.8 (CAr), 154.2 (C=O), 192.7 (C-S-); HRMS (ESI): m/z calcd for C25H23N5O4S 490.1549 [M + H]+, found 490.1544.
1-(4-Hydroxyphenyl)-2-((5-(2-((4-methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)ethanone (23): Prepared according to method C from 2-bromo-4’-hydroxyacetophenone (0.43 g). Yield 84%; white solid; m.p. 198–199 °C; IR (KBr) νmax (cm−1): 3380, 3300 (OH, NH), 1649 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.4 Hz, 2H, CH2C), 3.20 (q, J = 6.8, 13.0 Hz, 2H, CH2NH), 3.61 (s, 3H, CH3O), 4.76 (s, 2H, SCH2), 5.23 (t, J = 5.6 Hz, 1H, NH), 6.32 (d, J = 8.8 Hz, 2H, HAr2,6), 6.64 (d, J = 8.8 Hz, 2H, HAr3,5), 6.88 (d, J = 8.7 Hz, 2H, HAr’’), 7.46–7.61 (m, 5H, HAr’), 7.88 (d, J = 8.7 Hz, 2H, HAr’’), 10.58 (s, 1H, OH); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.7 (CH2C), 40.2 (CH2NH), 41.3 (SCH2), 55.3 (CH3O), 113.1, 114.6, 115.4, 126.7, 127.4, 129.9, 130.0, 131.1, 133.0, 142.2, 149.6, 150.8, 154.1 (CAr), 162.7 (C=O), 191.1 (C-S-); HRMS (ESI): m/z calcd for C25H24N4O3S 461.1647 [M + H]+, found 461.1653.
2-((5-(2-((4-Methoxyphenyl)amino)ethyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-1-(naphthalen-2-yl)ethanone (24): Prepared according to method C from 2-bromo-2′-acetonaphthone (0.50 g). Yield 91%; yellow solid; m.p. 154–155 °C; IR (KBr) νmax (cm−1): 3283 (NH); 1688 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.74 (t, J = 7.4 Hz, 2H, CH2C), 3.19 (q, J = 6.8, 14.0 Hz, 2H, CH2NH), 3.61 (s, 3H, CH3O), 5.01 (s, 2H, SCH2), 5.23 (t, J = 5.7 Hz, 1H, NH), 6.31 (d, J = 8.8 Hz, 2H, HAr2,6), 6.63 (d, J = 8.8 Hz, 2H, HAr3,5), 7.47–8.76 (m, 12H, HAr’, Ar’’); 13C-NMR (DMSO-d6, 101 MHz) δ: 24.7 (CH2C), 40.3 (CH2NH), 41.3 (SCH2), 55.3 (CH3O), 113.1, 114.6, 123.7, 127.1, 127.4, 127.7, 128.4, 128.9, 129.7; 130.0, 130.1, 130.6, 132.1, 132.6, 133.0, 142.2, 149.4, 150.8 (CAr), 154.1 (C=O), 193.1 (C-S-); (HRMS (ESI): m/z calcd for C29H26N4O2S 495.1854 [M + H]+, found 495.1849.
N-(4-Methoxyphenyl)-N-(2-(5-((2-oxo-2-(p-tolyl)ethyl)thio)-4-phenyl-4H-1,2,4-triazol-3-yl)ethyl)acetamide (25): A mixture of triazolethione 18 (0.2 g, 0.44 mmol) and acetyl chloride (10 mL) was heated at reflux for 1 h. The reaction mixture was poured into a water-ice mixture. Precipitate formed was filtered off, washed with water, and recrystallized from propan-2-ol. Yield 96%; white solid; m.p. 163–164 °C; IR (KBr) νmax (cm−1): 1651, 1510 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.61 (s, 3H, CH3); 2.38 (s, 3H, CH3), 2.82 (t, J = 6.8 Hz, 2H, CH2C), 3.66 (t, J = 6.8 Hz, 2H, CH2N); 3.77 (s, 3H, CH3O), 4.92 (s, 2H, SCH2), 6.94 (d, J = 8.8 Hz, 2H, HAr2,6), 7.08 (d, J = 8.8 Hz, 2H, HAr3,5), 7.35–7.43 (m, 4H, HAr’, Ar’’), 7.56–7.63 (m, 3H, HAr’), 7.91 (d, J = 8.0 Hz, 2H, HAr’’); 13C-NMR (DMSO-d6, 101 MHz) δ: 21.3 (CH3), 22.3 (CH3), 23.2 (CH2C), 40.5 (SCH2), 46.2 (CH2N), 55.4 (CH3O), 114.8, 127.2, 128.6, 129.4, 130.2, 130.6, 132.1, 132.7, 135.2, 144.5, 150.8, 153.5 (CAr), 158.5, 169.6 (C=O), 192.4 (C-S-); HRMS (ESI): m/z calcd for C28H28N4O3S 501.1960 [M + H]+, found 501.1963.
N-(2,5-Dimethyl-1H-pyrrol-1-yl)-3-((4-methoxyphenyl)amino)propanamide (26): A mixture of propanehydrazide 1 (1.05 g, 5 mmol), propan-2-ol (75 mL), hexane-2,5-dione (0.69 g, 6 mmol), and acetic acid (1.5 mL) was heated at reflux for 20 h. Afterwards cold water (25 mL) was added. Precipitate formed was filtered off and recrystallized from ethanol. Yield 93%; white solid; m.p. 119–120 °C; IR (KBr) νmax (cm−1): 3346, 2843 (NH), 1663 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.98, 2.02 (2s, 6H, 2CH3), 2.54 (t, J = 6.9 Hz, 2H, CH2CO), 3.32 (t, J = 6.9 Hz, 2H, CH2NH), 3.65 (s, 3H, CH3O), 5.23 (s, 1H, NH), 5.63, 5.71 (2s, 2H, 2CH), 6.54–6.78 (m, 4H, HAr), 10.60 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 10.9 (СH3), 33.3 (CH2CO), 40.1 (CH2NH), 55.2 (CH3O), 102.8, 113.3, 114.6, 126.7, 142.5, 150.9 (CAr), 170.4 (C=O); HRMS (ESI): m/z calcd for C16H21N3O2 289.1790 [M + H]+, found 289.1790.
3.2.5. General Procedure for the Synthesis of Compounds 27a,b
To propanehydrazide 1 (2.09 g, 10 mmol) dissolved in methanol (30 mL), the corresponding isatin (11 mmol) dissolved in methanol (15 mL) and glacial acetic acid (2–3 drops) were added. The reaction mixture was heated at reflux for 10–15 min until precipitate formed. The precipitate was filtered off while hot and recrystallized from DMF/H2O mixture.
3-((4-Methoxyphenyl)amino)-N’-(2-oxoindolin-3-ylidene)propanehydrazide (27a): Prepared from isatin (1.62 g). Yield 72%; yellow solid; m.p. 203–204 °C; IR (KBr) νmax (cm−1): 1694, 1728 (C=O), 3389, 3342, 3262 (NH); 1H-NMR (DMSO-d6, 400 MHz) δ: 3.01–3.34 (m, 4H, CH2CO+ CH2NH), 3.63 (s, 3H, CH3O), 5.26 (s, 1H, NHAr), 6.55–7.48 (m, 8H, HAr, Ar‘), 11.23 (s, 1H, NHC), 12.54 (s, 0.7H, NHCO), 12.98 (s, 0.3H, NHCO); 13C-NMR (DMSO-d6, 101 MHz) δ: 31.2 (CH2CO), 40.2 (CH2NH), 55.3 (CH3O), 111.1, 113.3, 114.6, 118.8, 119.8, 122.5, 131.4, 133.9, 142.3, 142.7, 150.9 (CAr), 162.5, 173.9 (C=O); HRMS (ESI): m/z calcd for C18H18N4O3 339.1457 [M + H]+, found 339.1467.
N’-(1-Benzyl-2-oxoindolin-3-ylidene)-3-((4-methoxyphenyl)amino)propanehydrazide (27b): Prepared from N-benzylisatin (2.61 g). Yield 78%; yellow solid; m.p. 123–124 °C; IR (KBr) νmax (cm−1): 1678, 1614 (C=O), 3384, 3219 (NH); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.68–3.43 (m, 4H, CH2CO + CH2NH), 3.64 (s, 3H, CH3O), 4.90–4.98 (m, 2H, CH2), 5.82 (s, 1H, NHAr), 6.60–7.61 (m, 13H, HAr, Ar′, Ar″), 11.29, 12.48 (2s, 0.7H, NHCO), 12.91 (s, 0.3H, NHCO); 13C-NMR (DMSO-d6, 101 MHz) δ: 31.1 (CH2CO), 39.9 (CH2), 42.5 (CH2NH), 55.3 (CH3O), 110.4, 113.8, 114.6, 114.8, 119.3, 120.2, 123.2, 127.4, 127.6, 128.7, 131.3, 135.7, 142.0, 142.5, 151.3 (CAr), 160.6, 173.8 (C=O); HRMS (ESI): m/z calcd for C25H24N4O3 429.1926 [M + H]+, found 429.1929.
3.2.6. General Procedure for the Synthesis of Compounds 28–37
To propanehydrazide 1 (0.63 g, 3 mmol) dissolved in methanol (20 mL), corresponding acetophenone (3 mmol) and acetic acid (4 drops; in the case of compounds 29, 30, 32, 33, and 36) were added. The reaction mixture was heated at 95 °C for 4–24 h. Precipitate formed was filtered off, washed with water, and recrystallized from methanol.
N’-(1-(Furan-2-yl)ethylidene)-3-((4-methoxyphenyl)amino)propanehydrazide (28): Prepared from 2-acetylfuran (0.33 g, 0.30 mL) by heating the reaction mixture for 22 h. Yield 76%; white solid; m.p. 142–143 °C; IR (KBr) νmax (cm−1): 3361, 3389 (NH), 1679 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.16 (s, 3H, CH3), 2.56 (t, J = 7.2 Hz, 0.8H, CH2CO), 2.85 (t, J = 7.2 Hz, 1.2H, CH2CO), 3.26 (t, J = 7.2 Hz, 2H, CH2NH), 3.63 (s, 3H, CH3O), 5.15, 5.20 (2t, J = 4.8 Hz, 1H, NHAr), 6.53–6.89 (m, 7H, HAr, furan), 10.30 (s, 0.4H, NH), 10.40 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.2, 13.5 (CH3), 32.6, 34.1 (CH2CO), 48.6 (CH2NH), 55.3 (CH3O), 110.6, 111.8, 113.1, 114.6, 139.6, 142.8, 144.2, 150.7 (CAr), 151.9 (C=N), 173.7 (C=O); HRMS (ESI): m/z calcd for C16H19N3O3 302.1504 [M + H]+, found 302.1501.
3-((4-Methoxyphenyl)amino)-N’-(1-(thiophen-2-yl)ethylidene)propanehydrazide (29). (Z/E isomeric mixture, 60 : 40%): Prepared from 2-acetylthiophene (0.38 g, 0.32 mL) by heating the reaction mixture for 12 h. Yield 69%; white solid; m.p. 160–162 °C; IR (KBr) νmax (cm−1): 3363, 3389 (NH), 1679 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.26 (s, 3H, CH3), 2.56 (t, J = 8.0 Hz, 0.8H, CH2CO), 2.86 (t, J = 8.0 Hz, 1.2H, CH2CO), 3.28 (t, J = 8.0 Hz, 2H, CH2NH), 3.63 (s, 3H, CH3O), 5.17 (s, 1H, NH), 6.55–6.74 (m, 4H, HAr), 7.05–7.07 (m, 1H, CH), 7.40–7.56 (m, 2H, CH), 10.36 (s, 0.4H, NH), 10.51 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.9, 14.4 (CH3), 32.6, 34.1 (CH2CO), 40.2 (CH2NH), 55.3 (CH3O), 113.2, 113.4, 114.6, 127.1, 127.4, 127.6, 127.7, 128.0, 128.6 (Cthiophene), 142.8, 142.9, 143.5, 143.6, 143.9, 147.9, 150.7, 150.9 (CAr), 167.6 (C=N), 173.6 (C=O); HRMS (ESI): m/z calcd for C16H19N3O2S 318.1276 [M + H]+, found 318.1273.
3-((4-Methoxyphenyl)amino)-N’-(1-(thiophen-2-yl)propylidene)propanehydrazide (30). (Z/E isomeric mixture, 60 : 40%): Prepared from 2-propionylthiophene (0.42 g, 0.37 mL) by heating the reaction mixture for 19 h. Yield 86%); white solid; m.p. 143–145 °C; IR (KBr) νmax (cm−1): 3345, 3390 (NH), 1675 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.05 (t, 3H, J = 8.0 Hz, CH3CH2), 2.58 (t, 0.8H, J = 8.0 Hz, CH2CO), 2.77 (qui, 2H, J = 8.0 Hz, CH3CH2), 2.87 (t, 1.2H, Hz, CH2CO), 3.28 (qui, 2H, J = 8.0 Hz, CH2NH), 3.63, 3.64 (2s, 3H, CH3O), 5.17, 5.18 (2s, 1H, NH), 6.55–6.75 (m, 4H, HAr), 7.04 (t, 1H, J = 4.0 Hz, CH), 7.39–7.56 (m, 2H, CH), 10.42 (s, 0.4H, NH), 10.65 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 11.1 (CH3CH2), 20.1, 20.4 (CH3CH2), 32.7, 34.1 (CH2CO), 40.3 (CH2NH), 55.3 (CH3O), 113.2, 113.4, 114.6, 126.6, 127.2, 127.5, 127.7, 127.9, 128.5 (Cthiophene), 142.7, 142.8, 142.8, 142.9, 148.3, 150.7, 150.9, 151.7 (CAr), 167.7 (C=N), 173.7 (C=O); HRMS (ESI): m/z calcd for C17H21N3O2S 332.1432 [M + H]+, found 332.1431.
3-((4-Methoxyphenyl)amino)-N’-(1-phenylethylidene)propanehydrazide (31): Prepared from acetophenone (0.36 g, 0.35 mL) by heating the reaction mixture for 4h. Yield 75%; white solid; m.p. 155–156 °C; IR (KBr) νmax (cm−1): 3385, 3358 (NH), 1680 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.25 (s, 3H, CH3), 2.59 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.93 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.24–3.32 (m, 2H, CH2NH), 3.63, 3.64 (2s, 3H, CH3O), 5.20 (s, 1H, NH), 6.52–6.77 (m, 4H, HAr), 7.33–7.82 (m, 5H, Hacetophenone), 10.39 (s, 0.4H, NH), 10.51 (s, 0.6H, NH), 13C-NMR (DMSO-d6, 101 MHz) δ: 13.6, 14.1 (CH3), 32.7, 34.1 (CH2CO), 40.3 (CH2NH), 55.3 (CH3O), 113.2, 113.4, 114.6, 126.0, 126.3, 128.3, 128.3, 128.9, 129.1 (Cacetophenone), 138.3, 142.8, 147.3, 150.7, 150.9 (CAr), 167.9 (C=N), 173.9 (C=O); HRMS (ESI): m/z calcd for C18H21N3O2 312.1712 [M + H]+, found 312.1710.
3-((4-Methoxyphenyl)amino)-N’-(1-phenylpropylidene)propanehydrazide (32) (Z/E isomeric mixture, 60 : 40%): Prepared from propiophenone (0.4 g, 0.39 mL) by heating the reaction mixture for 12 h. Yield 75%; white solid; m.p. 136–138 °C; IR (KBr) νmax (cm−1): 3345, 3395 (NH), 1676 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.01 (t, J = 8.0 Hz, 3H, CH3CH2), 2.61 (t, J = 8.0 Hz, 0.8H, CH2CO), 2.75–2.83 (m, 2H, CH3CH2), 2.95 (t, J = 8.0 Hz, 1.2H, CH2CO), 3.25–3.33 (m, 2H, CH2NH), 3.63, 3.64 (CH3O), 5.20 (s, 1H, NH), 6.51–6.78 (m, 4H, HAr), 7.34–7.81 (m, 5H, Hacetophenone), 10.49 (s, 0.4H, NH), 10.65 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 10.8 (CH3CH2), 19.2, 19.5 (CH3CH2), 32.7, 34.2 (CH2CO), 40.3 (CH2NH), 55.3 (CH3O), 113.2, 113.4, 114.6, 126.1, 126.4, 128.4, 128.5, 128.9, 129.0 (Cacetophenone), 137.1, 142.8, 142.9, 150.7, 150.9, 151.5, 154.8 (CAr), 168.1 (C=N), 174.1 (C=O); HRMS (ESI): m/z calcd for C19H23N3O2 326.1868 [M + H]+, found 326.1864.
N’-(1-(4-Aminophenyl)ethylidene)-3-((4-methoxyphenyl)amino)propanehydrazide (33) (Z/E isomeric mixture, 60 : 40%): Prepared from 4′-aminoacetophenone (0.41 g) by heating the reaction mixture for 24 h. Yield 68%; white solid; m.p. 137–138 °C; IR (KBr) νmax (cm−1): 3464, 3363, 3332 (NH, NH2), 1660 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.13 (s, 3H, CH3), 2.54 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.89 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.26 (t, J = 6.8 Hz, 2H, CH2NH), 3.64 (s, 3H, CH3O), 5.11–5.22 (m, 1H, NH), 5.42 (s, 2H, NH2), 6.52–6.59 (m, 4H, HAr), 6.68–6.75 (m, 2H, Hacetophenone), 7.45–7.53 (m, 2H, Hacetophenone), 10.14 (s, 0.4H, NH), 10.21 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.2, 13.7 (CH3), 32.7, 34.2 (CH2CO), 39.7, 40.2 (CH2NH), 55.3 (CH3O), 113.1, 113.2, 113.3, 114.6, 125.3, 125.5, 127.1, 127.5 (Cacetophenone), 142.9, 148.2, 149.9, 150.1, 150.7, 150.8 (Cacetophenone), 152.3 (CAr), 167.3 (C=N), 173.4 (C=O); HRMS (ESI): m/z calcd for C18H22N4O2 327.1821 [M + H]+, found 327.1842.
N’-(1-(3-Aminophenyl)ethylidene)-3-((4-methoxyphenyl)amino)propanehydrazide (34): Prepared from 3′-aminoacetophenone (0.41 g) by heating the reaction mixture for 24 h. Yield 71%; white solid; m.p. 109–110 °C; IR (KBr) νmax (cm−1): 3464, 3379, 3358 (NH, NH2), 1679 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.17 (s, 3H, CH3), 2.57 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.91 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.28 (t, J = 6.8 Hz, 2H, CH2NH), 3.63, 3.64 (2s, 3H, CH3O), 5.13 (s, 3H, NH+NH2), 6.51–7.08 (m, 8H, HAr, acetophenone), 10.27 (s, 0.4H, NH), 10.39 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.8, 14.2 (CH3), 32.8, 34.2 (CH2CO), 40.2, 40.3 (CH2NH), 55.3, 55.3 (CH3O), 111.5 (C-2acetophenone), 113.2, 113.4, 113.9 (C-4acetophenone), 114.3, 114.7, 115.0 (C-6acetophenone), 128.7, 128.7, 138.9, 139.0 (C-5acetophenone), 142.9 (C-1acetophenone), 148.2, 148.5, 148.6 (C-3acetophenone), 150.7, 150.9, 151.8 (CAr), 167.8 (C=N), 173.8 (C=O); HRMS (ESI): m/z calcd for C18H22N4O2 327.1821 [M + H]+, found 327.1835.
N’-(1-(3,4-Dimethoxyphenyl)ethylidene)-3-((4-methoxyphenyl)amino)propanehydrazide (35): Prepared from 3′,4′-dimethoxyacetophenone (0.54 g) by heating the reaction mixture for 48 h. Yield 27%; white solid; m.p. 73–74 °C; IR (KBr) νmax (cm−1): 3465, 3366 (NH), 1675 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.21, 2.22 (2s, 3H, CH3), 2.57 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.92 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.24–3.32 (m, 2H, CH2NH), 3.62, 3.64 (2s, 3H, CH3O), 3.72, 3.78, 3.81, 3.84 (4s, 6H, 2CH3O), 5.15–5.23 (m, 1H, NH), 6.50–6.75 (m, 4H, HAr), 6.92–7.65 (m, 3H, Hacetophenone), 10.41 (s, 1H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.5, 14.0 (CH3), 32.8, 34.2 (CH2CO), 39.6, 40.4 (CH2NH), 55.2, 55.3, 55.3, 55.4, 55.5, 55.7 (CH3O), 109.1, 109.2, 110.2, 110.8, 111.0, 111.2 (C-2,5acetophenone), 113.2, 113.3, 114.6, 114.6, 119.2, 119.6 (C-6acetophenone), 129.9, 130.9, 131.0 (C-1acetophenone), 142.9, 148.4, 148.5, 148.5 (C-3acetophenone), 149.8, 150.0, 150.7, 150.8, 151.3, 153.0 (C-4acetophenone) (CAr), 167.7 (C=N), 173.8 (C=O); HRMS (ESI): m/z calcd for C20H25N3O4 372.1923 [M + H]+, found 372.1921.
3-((4-Methoxyphenyl)amino)-N’-(1-(naphthalen-1-yl)ethylidene)propanehydrazide (36). (Z/E isomeric mixture, 60 : 40%): Prepared from 1-acetonaphthone (0.51 g, 0.46 mL) by heating the reaction mixture for 5 h. Yield 65%; white solid; m.p. 152–154 °C; IR KBr) νmax (cm−1): 3337, 3290 (NH), 1667 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.37, 2.38 (2s, 3H, CH3), 2.64 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.79 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.19–3.32 (m, 2H, CH2NH), 3.57, 3.65 (2s, 3H, CH3O), 5.15 (s, 0.6H, NH), 5.22 (s, 0.4H, NH), 6.43–6.76 (m, 4H, HAr), 7.48–7.58 (m, 4H, Hacetonaphthone), 7.90–8.21 (m, 3H, Hacetonaphthone), 10.51 (s, 0.4H, NH), 10.61 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 18.8, 19.1 (CH3), 32.9, 34.2 (CH2CO), 40.2 (CH2NH), 55.2, 55.3 (CH3O), 113.1, 113.4, 114.4, 114.7, 125.2, 125.3, 125.3, 125.6, 125.9, 126.0, 126.5, 126.6, 128.4, 128.6, 128.7 (Cacetonaphthone), 130.2, 133.4, 137.7, 137.9, 142.6, 142.9, 149.4, 150.6, 150.9 (CAr), 153.5 (C=N), 167.9, 173.9 (C=O); HRMS (ESI): m/z calcd for C22H23N3O2 362.1868 [M + H]+, found 362.1868.
3-((4-Methoxyphenyl)amino)-N’-(1-(naphthalen-1-yl)ethylidene)propanehydrazide (36). (Z/E isomeric mixture, 60 : 40%): Prepared from 1-acetonaphthone (0.51 g, 0.46 mL) by heating the reaction mixture for 5 h. Yield 65%; white solid; m.p. 152–154 °C; IR KBr) νmax (cm−1): 3337, 3290 (NH), 1667 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.37, 2.38 (2s, 3H, CH3), 2.64 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.79 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.19–3.32 (m, 2H, CH2NH), 3.57, 3.65 (2s, 3H, CH3O), 5.15 (s, 0.6H, NH), 5.22 (s, 0.4H, NH), 6.43–6.76 (m, 4H, HAr), 7.48–7.58 (m, 4H, Hacetonaphthone), 7.90–8.21 (m, 3H, Hacetonaphthone), 10.51 (s, 0.4H, NH), 10.61 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 18.8, 19.1 (CH3), 32.9, 34.2 (CH2CO), 40.2 (CH2NH), 55.2, 55.3 (CH3O), 113.1, 113.4, 114.4, 114.7, 125.2, 125.3, 125.3, 125.6, 125.9, 126.0, 126.5, 126.6, 128.4, 128.6, 128.7 (Cacetonaphthone), 130.2, 133.4, 137.7, 137.9, 142.6, 142.9, 149.4, 150.6, 150.9 (CAr), 153.5 (C=N), 167.9, 173.9 (C=O); HRMS (ESI): m/z calcd for C22H23N3O2 362.1868 [M + H]+, found 362.1868.
3-((4-Methoxyphenyl)amino)-N’-(1-(naphthalen-2-yl)ethylidene)propanehydrazide (37): Prepared from 2-acetonaphthone (0.51 g) by heating the reaction mixture for 16 h. Yield 69%; white solid; m.p. 141–142 °C; IR (KBr) νmax (cm−1): 3367, 3310 (NH) 1662 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.37 (s, 3H, CH3), 2.64 (t, J = 6.8 Hz, 0.8H, CH2CO), 3.01 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.27–3.39 (m, 2H, CH2NH), 3.61, 3.64 (2s, 3H, CH3O), 5.23 (s, 1H, NH), 6.53–6.78 (m, 4H, HAr), 7.48–8.27 (m, 7H, Hacetonaphthone), 10.50 (s, 0.4H, NH), 10.62 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.4, 13.8 (CH3), 32.8, 34.3 (CH2CO), 39.6, 40.3 (CH2NH); 55.2, 55.3 (CH3O), 113.2, 113.4, 114.6, 123.4, 123.7, 125.9, 126.2, 126.4, 126.7, 126.7, 127.5, 127.6, 127.7, 128.4, 132.8, 132.8, 133.1, 133.2, 135.6, 135.7 (Cacetonaphthone); 142.9, 142.9, 150.6, 150.7, 150.9 (CAr), 153.5 (C=N), 168.1, 174.0 (C=O); HRMS (ESI): m/z calcd for C22H23N3O2 362.1868 [M + H]+, found 362.1868.
N-(4-Methoxyphenyl)-N-(3-oxo-3-(2-(1-phenylethylidene)hydrazinyl)propyl)acetamide (38): To propanehydrazide 31 (0.19 g, 0.6 mmol) dissolved in methanol (5 mL), acetic anhydride (5 mL) was added and the reaction mixture was heated at reflux for 2 h. Afterwards water (10 mL) was added. Precipitate formed was filtered off, washed with methanol, and recrystallized from methanol. Yield 59 %; white solid; m.p. 145–146 °C; IR (KBr) νmax (cm−1): 3176 (NH) 1658; 1607 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 1.70 (s, 3H, CH3), 2.19, 2.23 (2s, 3H, CH3), 2.54 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.84 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.77 (s, 3H, CH3O), 3.87 (t, 2H, CH2N), 6.93–7.78 (m, 9H, HAr), 10.37 (s, 0.4H, NH), 10.47 (s, 0.6H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 13.4, 14.1 (CH3), 22.5 (CH3), 31.5, 32.8 (CH2CO), 44.9, 45.1 (CH2N), 55.3 (CH3O), 114.7, 125.9, 126.3, 128.3, 128.9, 129.4 (Cacetophenone), 135.4, 138.0, 138.3, 147.2, 158.4 (CAr), 167.1, 169.2, 169.2, 173.1 (C=O); HRMS (ESI): m/z calcd for C20H23N3O3 354.1817 [M + H]+, found 354.1844.
N-(1,3-Dioxoisoindolin-2-yl)-3-((4-methoxyphenyl)amino)propanamide (39): To propanehidrazyde 1 (0.63 g, 3 mmol) dissolved in 1,4-dioxane (20 mL), phthalic anhydride (0.89 g, 6 mmol) was added and the reaction mixture was heated at reflux for 4 h. Afterwards Na2CO3 was added until pH 7. Precipitate formed was filtered off, washed with water, and recrystallized from methanol. Yield 81%; white solid; m.p. 147–148 °C; IR (KBr) νmax (cm−1): 3331, 3290 (NH), 1664 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ: 2.46 (t, J = 6.8 Hz, 0.8H, CH2CO), 2.89 (t, J = 6.8 Hz, 1.2H, CH2CO), 3.25–3.30 (m, 2H, CH2NH), 3.64 (s, 3H, CH3O), 5.14–5.24 (m, 1H, NH), 6.54–6.75 (m, 4H, HAr), 7.37–7.70 (m, 4H, Hphthal), 11.32 (s, 0.6H, NH), 11.38 (s, 0.4H, NH); 13C-NMR (DMSO-d6, 101 MHz) δ: 32.2 (CH2CO), 34.3 (CH2NH); 55.3, 55.3 (CH3O), 113.2, 113.2, 114.6, 126.6, 126.9, 128.8, 129.7, 129.9, 134.3, 134.3, 142.7, 150.7 (CAr), 167.3, 173.1 (C=O); HRMS (ESI): m/z calcd for C18H17N3O4 340.1297 [M + H]+, found 340.1299.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}