Thiabendazole and Thiabendazole-Formic Acid Solvate: A Computational, Crystallographic, Spectroscopic and Thermal Study


 ,
,  , , ,
, , ,  and
and 
Abstract
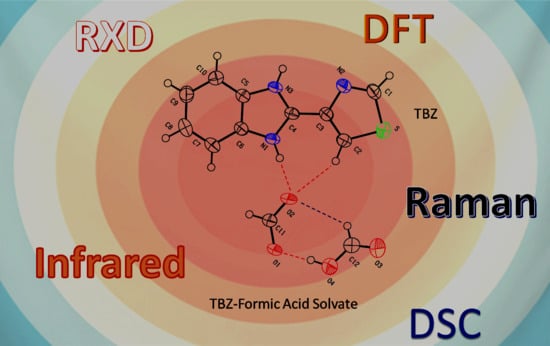
:
1. Introduction
2. Materials and Methods
2.1. Experimental Details
2.2. Computational Methods
3. Results and Discussion
3.1. DFT Studies on the Isolated Molecule of Thiabendazole
3.1.1. Structural Details
3.1.2. Spectroscopic Properties
3.2. DFT Studies on the Isolated Thiabendazole-Formic Acid Complex
3.3. Single Crystal X-Ray Diffraction Studies on TBZ-Formic Acid Solvate, Theoretical Predictions, and Comparison with the Crystal of Pure TBZ
3.4. Hirshfeld Analysis of Crystalline TBZ and TBZ-Formic Acid Solvate
3.5. DSC Study of TBZ and TBZ-Formic Acid Solvate Crystalline Materials
3.6. Infrared and Raman Spectra of TBZ and TBZ-Formic Acid Solvate Crystalline Materials
3.6.1. Infrared Spectra
3.6.2. Raman Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Brown, H.D.; Matzuk, A.R.; Ilves, I.R.; Peterson, L.H.; Harris, S.A.; Sarett, L.H.; Egerton, J.R.; Yakstis, J.J.; Campbell, W.C.; Cuckler, A.C. Antiparasitic drugs. iv. 2-(4’-thiazolyl)-benzimidazole, a new anthelmintic. J. Am. Chem. Soc. 1961, 83, 1764–1765. [Google Scholar] [CrossRef]
- Robinson, H.J.; Phares, H.F.; Graessle, O.E. Antimycotic properties of thiabendazole. J. Investig. Dermatol. 1964, 42, 479–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.E.; Dolin, R.; Blaser, M.J. Drugs for Helminths. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Benett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 1. [Google Scholar]
- Robinson, H.J.; Stoerk, H.C.; Graessle, O.E. Studies on the toxicologic and pharmacologic properties of thiabendazole. Toxicol. Appl. Pharmacol. 1965, 7, 53–63. [Google Scholar] [CrossRef]
- Feng, J.; Hu, Y.; Grant, E.; Lu, X. Determination of thiabendazole in orange juice using an MISPE-SERS chemosensor. Food Chem. 2018, 239, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.J.D.V.; Toledo, M.C.D.F. Resíduos dos fungicidas tiabendazol e imazalil em laranjas pêra (Citrus sinensis (l.) Osbech cv. Pêra)*. Pestic. Rev. de Ecotoxicologia e Meio Ambient. 1999, 9, 125–136. [Google Scholar] [CrossRef]
- Fripp, I.J.; Dettman, E.B. Thiabendazole as a postharvest treatment against Sclerotinia fruticola in dessert peaches. Aust. J. Exp. Agric. Anim. Husb. 1969, 9, 9–11. [Google Scholar] [CrossRef]
- Zhang, C.; Zhong, B.; Yang, S.; Pan, L.; Yu, S.; Li, Z.; Li, S.; Su, B.; Meng, X. Synthesis and biological evaluation of thiabendazole derivatives as anti-angiogenesis and vascular disrupting agents. Bioorganic Med. Chem. 2015, 23, 3774–3780. [Google Scholar] [CrossRef]
- Grevy, J.M.; Tellez, F.; Bernès, S.; Noth, H.; Contreras, R.; Barba-Behrens, N. Coordination compounds of thiabendazole with main group and transition metal ions. Inorganica Chim. Acta 2002, 339, 532–542. [Google Scholar] [CrossRef]
- Igual-Adell, R.; Oltra-Alcaraz, C.; Soler-Company, E.; Sánchez-Sánchez, P.; Matogo-Oyana, J.; Rodríguez-Calabuig, D. Efficacy and safety of ivermectin and thiabendazole in the treatment of strongyloidiasis. Expert Opin. Pharmacother. 2004, 5, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.; Burton, D.R.; Caria, S.; Desbois, S.; Gee, C.L.; Fazio, V.; Kvansakul, M.; Marshall, B.; Mills, G.; Richter, V.; et al. Crystallization reports are the backbone of Acta Cryst. F, but do they have any spine? Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2013, 69, 712–718. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Kim, M.-K.; Lee, C.-J.; Jung, Y.-M.; Lee, M.S. Surface-enhanced Raman Spectroscopy of Benzimidazolic Fungicides: Benzimidazole and Thiabendazole. Bull. Korean Chem. Soc. 2009, 30, 2930–2934. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, Q.; Li, L. PLS model investigation of thiabendazole based on THz spectrum. J. Quant. Spectrosc. Radiat. Transf. 2013, 117, 7–14. [Google Scholar] [CrossRef]
- Silva, M.; Gonring, K.; Silva, R.; Fonseca, M.; Borges, M.; Nunes, O.; Forim, M.; Borges, K.B.; Borges, W.D.S. Fourier transform infrared spectroscopy, thermogravimetric analysis, scanning electron microscopy as supporting tools in quality control of antiparasitics. Química Nova 2017, 41, 258–267. [Google Scholar] [CrossRef]
- Lombardi, B.M.; Sánchez, R.M.T.; Eloy, P.; Genet, M. Interaction of thiabendazole and benzimidazole with montmorillonite. Appl. Clay Sci. 2006, 33, 59–65. [Google Scholar] [CrossRef]
- Wei, S.-Q.; Lin, C.; Yin, X.-H.; Huang, Y.-J.; Luo, P.-Q. Hydrothermal Synthesis, Crystal Structure of Four Novel Complexes Based on Thiabendazole Ligand. Bull. Korean Chem. Soc. 2012, 33, 2917–2924. [Google Scholar] [CrossRef] [Green Version]
- Grant, D.J.; York, P. Entropy of processing: A new quantity for comparing the solid state disorder of pharmaceutical materials. Int. J. Pharm. 1986, 30, 161–180. [Google Scholar] [CrossRef]
- Clarke, H.D.; Arora, K.K.; Bass, H.; Kavuru, P.; Ong, T.T.; Pujari, T.; Wojtas, L.; Zaworotko, M.J. Structure−Stability Relationships in Cocrystal Hydrates: Does the Promiscuity of Water Make Crystalline Hydrates the Nemesis of Crystal Engineering? Cryst. Growth Des. 2010, 10, 2152–2167. [Google Scholar] [CrossRef]
- Takieddin, K.; Khimyak, Y.Z.; Fabian, L. Prediction of Hydrate and Solvate Formation Using Statistical Models. Cryst. Growth Des. 2015, 16, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Flores-Alamo, M.; González-Martínez, S.; Blum, S.E.C. 2-(1,3-Thia-zol-4-yl)benzimidazolium nitrate monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o812. [Google Scholar] [CrossRef] [Green Version]
- Bruker APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Platon, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2020. [Google Scholar]
- Macrae, C.; Edgington, P.R.; McCabe, P.E.; Pidcock, E.; Shields, G.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8; Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com. (accessed on 9 June 2020).
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, 1st ed.; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Frisch, M.J.; Scuseria, G.E. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- O’Neil, M.J. TheMerck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 14th ed.; Merck: Whitehouse Station, NJ, USA, 2006; pp. 322–323. [Google Scholar]
- Rajzman, A. Determination of thiabendazole in citrus fruits by ultraviolet spectrophotometry. Analyst 1974, 99, 120. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, 17.5; University of Western Australia: Crawley, Australia, 2013. [Google Scholar]
- McKinnon, J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Chem.-A Eur. J. 1998, 4, 2136–2141. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Rohl, A.L.; Moret, M.; Kaminsky, W.; Claborn, K.; McKinnon, J.; Kahr, B. Hirshfeld Surfaces Identify Inadequacies in Computations of Intermolecular Interactions in Crystals: Pentamorphic 1,8-Dihydroxyanthraquinone. Cryst. Growth Des. 2008, 8, 4517–4525. [Google Scholar] [CrossRef]
- Parkin, A.; Barr, G.; Dong, W.; Gilmore, C.J.; Jayatilaka, D.; McKinnon, J.J.; Spackman, M.A.; Wilson, C.C. Comparing entire crystal structures: Structural genetic fingerprinting. CrystEngComm 2007, 9, 648–652. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency (EPA) Database, Thiabendazole, 148-79-8|DTXSID0021337. Available online: https://comptox.epa.gov/dashboard/DTXSID0021337 (accessed on 9 June 2020).
- Rozenberg, M.; Shoham, G.; Reva, I.; Fausto, R. Spontaneous self-association of adenine and uracil in polycrystals from low temperature FTIR spectra in the range below 1000 cm−1. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 62, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Rozenberg, M.; Shoham, G.; Reva, I.; Fausto, R. A correlation between the proton stretching vibration red shift and the hydrogen bond length in polycrystalline amino acids and peptides. Phys. Chem. Chem. Phys. 2005, 7, 2376–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenberg, M.; Shoham, G.; Reva, I.; Fausto, R. Low temperature FTIR spectroscopy and hydrogen bonding in cytosine polycrystals. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 463–470. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| trans | gauche | trans | gauche | ||
|---|---|---|---|---|---|
| C5-C6 | 1.415 | 1.412 | C6-C7-H7 | 120.4 | 120.3 |
| C6-C7 | 1.397 | 1.397 | C6-N1-C4 | 105.1 | 105.5 |
| C6-N1 | 1.383 | 1.383 | C10-C5-N3 | 132.9 | 132.9 |
| C5-C10 | 1.392 | 1.392 | C5-C10-C9 | 116.8 | 116.7 |
| C5-N3 | 1.378 | 1.381 | C5-C10-H10 | 121.9 | 122.1 |
| C10-C9 | 1.388 | 1.388 | C5-N3-H3 | 128.8 | 126.1 |
| C10-H10 | 1.081 | 1.081 | C5-N3-C4 | 107.0 | 107.0 |
| C8-C9 | 1.406 | 1.406 | C9-C10-H10 | 121.3 | 121.2 |
| C9-H9 | 1.081 | 1.081 | C10-C9-C8 | 121.5 | 121.5 |
| C8-C7 | 1.386 | 1.386 | C10-C9-H9 | 119.3 | 119.2 |
| C8-H8 | 1.081 | 1.081 | C8-C9-H9 | 119.2 | 119.2 |
| C7-H7 | 1.081 | 1.081 | C9-C8-C7 | 121.5 | 121.4 |
| N3-H3 | 1.006 | 1.004 | C9-C8-H8 | 119.0 | 119.0 |
| C4-N1 | 1.313 | 1.309 | C7-C8-H8 | 119.5 | 119.6 |
| C2-H2 | 1.075 | 1.076 | C8-C7-H7 | 121.6 | 121.7 |
| C2-C3 | 1.367 | 1.371 | C4-N3-H3 | 124.2 | 125.9 |
| C2-S | 1.720 | 1.721 | N1-C4-C3 | 126.1 | 125.9 |
| C1-H1A | 1.079 | 1.079 | N1-C4-N3 | 113.0 | 112.6 |
| C1-S | 1.738 | 1.744 | C3-C2-H2 | 127.7 | 128.8 |
| C1-N2 | 1.295 | 1.291 | S-C2-H2 | 122.5 | 120.8 |
| C3-C4 | 1.454 | 1.460 | C3-C2-S | 109.8 | 110.4 |
| C3-N2 | 1.380 | 1.378 | C2-C3-C4 | 125.7 | 125.3 |
| C4-N3 | 1.374 | 1.384 | C2-C3-N2 | 115.3 | 114.8 |
| C5-C6-C7 | 119.8 | 119.8 | C2-S-C1 | 89.2 | 88.5 |
| C5-C6-N1 | 110.2 | 110.3 | C1-S-C1-H1A | 121.0 | 120.6 |
| C6-C5-C10 | 122.4 | 122.5 | N2-C1-H1A | 124.2 | 124.2 |
| C6-C5-N3 | 104.6 | 104.6 | S-C1-N2 | 114.8 | 115.2 |
| C7-C6-N1 | 130.0 | 129.8 | C1-N2-C3 | 110.9 | 111.1 |
| C6-C7-C8 | 118.0 | 118.0 | C4-C3-N2 | 119.0 | 119.9 |
| C3-C4-N3 | 120.8 | 121.5 | N1-C4-C3-N2 | 180.0 | 21.1 |
| ν | IIR | Approximate b Description | ν | IIR | Approximate Description | ν | IIR | Approximate Description |
|---|---|---|---|---|---|---|---|---|
| 3573 | 79 | νN-H | 1226 | 3 | δCH Ph | 732 | 70 | γCH Ph |
| 3202 | 8 | νC2-H | 1218 | 6 | δC1-H | 721 | 2 | γ(inter-rings) |
| 3156 | 0 | νC1-H | 1167 | 6 | δN-H | 654 | 1 | τThi |
| 3131 | 14 | νCH Ph | 1146 | 4 | δCH Ph | 626 | 1 | δThi |
| 3123 | 18 | νCH Ph | 1113 | 2 | δCH Ph | 617 | 2 | δCCC Ph |
| 3112 | 7 | νCH Ph | 1073 | 2 | δC2-H | 579 | 4 | τPh |
| 3103 | 0 | νCH Ph | 1005 | 6 | νC-C Ph | 567 | 1 | δCCC Ph |
| 1620 | 5 | νC=C Ph | 968 | 3 | δNCN Imi | 529 | 11 | δ(inter-rings) |
| 1582 | 4 | νC=C Ph | 962 | 0 | γCH Ph | 509 | 80 | γN-H |
| 1560 | 8 | νC3-C4 | 923 | 2 | γCH Ph | 480 | 6 | τThi |
| 1495 | 34 | νC2-C3 | 892 | 15 | δCCC Ph | 428 | 1 | τPh |
| 1488 | 3 | δCH Ph | 888 | 8 | δC3N2C1 | 333 | 0 | δ(butterfly) |
| 1445 | 7 | δCH Ph | 857 | 38 | νC2-S | 331 | 1 | δ(inter-rings) |
| 1422 | 44 | νC1-N2 | 837 | 1 | γCH Ph | 282 | 0 | δ(skeletal) |
| 1392 | 41 | νC4-N3/δN-H | 820 | 40 | γC2H/γC1H | 251 | 3 | τImi |
| 1344 | 49 | νC=C Ph | 808 | 4 | δCCC Ph | 188 | 0 | τ(skeletal) |
| 1305 | 28 | νC3-N2 | 764 | 1 | νC1-S | 94 | 3 | δ(inter-rings) |
| 1296 | 16 | δCH Ph | 760 | 7 | γC2H/γC1H | 67 | 0 | τ(skeletal) |
| 1261 | 51 | νC6-N1/νC5-N3 | 754 | 7 | τPh | 57 | 4 | τC3-C4 |
| Excited State | Wavelength (nm) | Oscillator Strength (f) | Most Relevant One-Electron Transition a |
|---|---|---|---|
| T1 (A′) | 433.77 | 0 | HOMO→LUMO (0.6) |
| T2 (A′) | 364.25 | 0 | HOMO-1→LUMO (0.5) |
| T3 (A′) | 327.01 | 0 | HOMO→LUMO+1 (0.5) |
| S1 (A′) | 307.01 | 0.2636 | HOMO→LUMO (0.7) |
| S2 (A′) | 291.22 | 0.0542 | HOMO-1→LUMO (0.7) |
| T4 (A′) | 286.09 | 0 | HOMO-1→LUMO+4 (0.4); HOMO-1→LUMO+2 (0.3) |
| T5 (A′) | 280.58 | 0 | HOMO-2→LUMO (0.4) |
| S3 (A′) | 271.17 | 0.341 | HOMO→LUMO+1 (0.7) |
| T6 (A′) | 268.91 | 0 | HOMO→LUMO+4 (0.5) |
| S4 (A′) | 266.33 | 0.0694 | HOMO-1→LUMO+1 (0.6) |
| S5 (A″) | 255.47 | 0.0009 | HOMO→LUMO+2 (0.7) |
| S6 (A″) | 254.22 | 0.0008 | HOMO-3→LUMO+1 (0.7) |
| Exp. | B3LYP | B3LYP-D2 | 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| pob-TZVP | %E | 6-31G(d,p) | %E | pob-TZVP | %E | 6-31G(d,p) | %E | ||
| TBZ orthorhombic (Z = 8) Pbca | |||||||||
| a | 17.052 | 18.928 | 11.0 | 19.325 | 13.3 | 17.411 | 2.1 | 17.620 | 3.3 |
| b | 10.998 | 10.399 | −5.4 | 11.482 | 4.4 | 10.132 | −7.9 | 10.632 | −3.3 |
| c | 10.030 | 9.923 | −1.1 | 10.081 | 0.5 | 9.848 | −1.8 | 9.946 | −0.8 |
| Volume | 1881.01 | 1953.24 | 3.8 | 2236.91 | 18.9 | 1737.44 | −7.6 | 1863.16 | −0.9 |
| TBZ-formic acid solvate monoclinic (Z = 4) P21/c | |||||||||
| a | 3.8339 | 4.219 | 10.0 | 4.295 | 12.0 | 3.621 | −5.6 | 3.734 | −2.6 |
| b | 22.195 | 22.580 | 1.7 | 22.606 | 1.9 | 22.046 | −0.7 | 22.133 | −0.3 |
| c | 15.3695 | 14.442 | −6.0 | 15.168 | −1.3 | 15.157 | −1.4 | 15.181 | −1.2 |
| β | 93.412 | 100.542 | 7.6 | 92.51 | −1.0 | 97.596 | 4.5 | 95.477 | 2.2 |
| Volume | 1305.52 | 1352.61 | 3.6 | 1471.27 | 12.7 | 1199.27 | −8.1 | 1248.98 | −4.3 |
| νexp | νpred | Approximate b Description | νexp | νpred | Approximate b Description |
|---|---|---|---|---|---|
| 1622 | 1631 | νC=C Ph | 1158 | 1160 | δCH Ph |
| 1592 | 1595 | νC=C Ph | 1095 | 1109 | δC2-H |
| 1579 | 1577 | νC3-C4 | 1013 | 1018 | νC-C Ph |
| 1492 | 1500 | νC3-C2 | 986 | 983 | δNCN Imi |
| 1481 | 1490 | νC4-N3 | 927 | 927 | γCH Ph |
| 1455 | 1460 | δCH Ph | 902 | 946 | γN-H |
| 1412 | 1438 | νC1-N2 | 874 | 899 | δC3N2C1 |
| 1403 | 1422 | δN-H | 835 | 864 | νC2-S |
| 1358 | 1371 | νC=C Ph | 822 | 848/840 | γC2H/γC1H |
 | νC3-N2 | n.obs. | 815 | νC1-S/δCCC Ph | |
| δCH Ph | 780 | 767 | γC2H/γC1H | ||
| 1278 | 1285 | νC6-N1/νC5-N3 | 769 | 756 | γCH Ph |
| 1254 | 1271 | δN-H | 731 | 751 | γ (inter-rings) |
| 1231 | 1230 | δC10-H/δC7-H | 723 | 739 | γCH Ph |
| 1197 | 1202 | δC2-H/δC1-H | 653 | 654 | τThi |
| TBZ | TBZ-Formic Acid Solvate | ||||
|---|---|---|---|---|---|
| νexp | νpred | App. Description | νexp | νpred | App. Description |
| 3091 | 3100 | νC-H Thi | 3121 | 3126 | νC-H Thi |
| 3065 | 3063 | νC-H Ph | 3107 | 3116 | νC-H Thi |
| 3044 | 3043 | νC-H Ph | 3078 | 3080 | νC-H Ph |
| 2976 | 2967 | νN-H | 3068 | 3062 | νC-H Ph |
| b | 2912 | νN-H | 2921 | 2921 | νC-H HCOOH |
| 1623 | 1629 | νC=C Ph | b | 2889 | νN3-H |
| 1593 | 1593 | νC=C Ph | 2835 | 2835 | νC-H HCOO− |
| 1579 | 1574 | νC3-C4 | b | 2669 | νO-H HCOOH/νN-H |
| 1547 | 1564 | νC3-C4 | b | 2632 | νO-H HCOOH/νN-H |
| 1494 | 1501 | νC3-C2 | b | 2546 | νO-H HCOOH |
| 1482 | 1487 | νC4-N1 | b | 2514 | νN1-H |
| 1456 | 1461 | δCH Ph | 1732 | 1756 | νC=O HCOOH |
| 1425 | 1434 | νC1-N2 |  | δN1-H | |
| 1416 | 1419 | νC4-N3 | νC=C Ph | ||
| 1357 | 1369 | νC=C Ph | 1597 | 1597 | νC3-C4 |
| 1303 | 1317 | δCH Thi | 1544 | 1544 | δN1-H/δN3-H |
| 1303 | 1309 | δCH Ph | 1494 | 1503 | νC3-C2 |
| 1278 | 1278 | νC5-N3 | 1469 | 1479 | νC1-N2 |
| 1257 | 1273 | δN-H |  | δO-H HCOOH | |
| 1231 | 1227 | νC6-N1 | δCH Ph | ||
| 1201 | 1207 | δC2-H/δC1-H | 1428 | 1446 | νC4-N3 |
| 1157 | 1157 | δCH Ph | 1415 | 1439 | νC4-N1 |
| 1120 | 1119 | δCH Ph | 1387 | 1390 | δC-H HCOO− |
| 1097 | 1109 | δC2-H | 1375 | 1386 | δC-H HCOOH |
| 1014 | 1017 | νC-C Ph | 1359 | 1359 | νO-C-O HCOO− |
| 989 | 985 | δNCN Imi | 1324 | 1328 | νC3-N2 |
| 970 | 967 | γCH Ph | 1311 | 1310 | δCH Ph |
| 936 | 948 | γN-H | 1291 | 1300 | νC6-N1/νC-C Ph |
 | δC3N2C1 | 1263 | 1278 | νC5-N3 | |
| δCC Ph | 1225 | 1234 | νC-OH HCOOH | ||
| 879 | 867 | νC2-S | 1202 | 1206 | δC2-H/δC1-H |
| 858 | 844 | γC2H/γC1H | 1161 | 1168 | δCH Ph |
| 840 | 836 | γCH Ph | 1140 | 1133 | δC2-H |
| 823 | 814 | νC1-S/δCCC Ph | 1124 | 1124 | δCH Ph |
| 782 | 783 | γC2H/γC1H | 1052 | 1051 | γCH HCOO− |
| 771 | 770 | νC1-S/δCCC Ph | 1048 | 1041 | γOCO HCOOH |
| 749 | 761 | γCH Ph | 1011 | 1017 | νC-C Ph |
| 726 | 732 | γ(inter-rings) | 981 | 990 | γCH Ph/γN1H |
| 655 | 654 | τThi | 971 | 978 | δNCN Imi |
| 636 | 629 | δThi | 932 | 930 | γN3H |
| 626 | 622 | δCCC Ph | 901 | 896 | γCH Ph |
| 618 | 613 | δCCC Ph | 879 | 886 | δC3N2C1 |
| 578 | 575 | τPh | 842 | 849 | νC2-S |
| 577 | 569 | δCCC Ph |  | γCH Ph | |
| 538 | 539 | δ(inter-rings) | δN2C1S | ||
| 491 | 487 | τThi | 781 | 776 | γC2H/γC1H |
| 451 | 465 | τPh | 750 | 747 | γCH Ph |
| 357 | 363 | δ(butterfly) | 724 | 719 | γ (inter-rings) |
| 331 | 329 | δ(inter-rings) | 699 | 693 | δOCO HCOOH |
| 292 | 289 | δ(skeletal) |  | τThi | |
| 270 | 273 | τImi | δThi | ||
| 202 | 209 | τ (skeletal) | 609 | 614 | δCCC Ph |
| 570 | 567 | τPh | |||
| 530 | 535 | δCCC Ph | |||
| 488 | 487 | τThi | |||
| 425 | 432 | τPh | |||
| 354 | 344 | δ(butterfly) | |||
 | δ(inter-rings) | ||||
| νHCOOH…HCOO− | |||||
| 283 | 284 | δ(skeletal)/τImi | |||
| 198 | 221 | τHCOOH | |||
| 182 | 209 | τ(skeletal) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabanez, A.M.; Nogueira, B.A.; Milani, A.; Eusébio, M.E.S.; Paixão, J.A.; Nur Kabuk, H.; Jajuga, M.; Ildiz, G.O.; Fausto, R. Thiabendazole and Thiabendazole-Formic Acid Solvate: A Computational, Crystallographic, Spectroscopic and Thermal Study. Molecules 2020, 25, 3083. https://doi.org/10.3390/molecules25133083
Tabanez AM, Nogueira BA, Milani A, Eusébio MES, Paixão JA, Nur Kabuk H, Jajuga M, Ildiz GO, Fausto R. Thiabendazole and Thiabendazole-Formic Acid Solvate: A Computational, Crystallographic, Spectroscopic and Thermal Study. Molecules. 2020; 25(13):3083. https://doi.org/10.3390/molecules25133083
Chicago/Turabian StyleTabanez, Andreia M., Bernardo A. Nogueira, Alberto Milani, M. Ermelinda S. Eusébio, José A. Paixão, Hayrunnisa Nur Kabuk, Maria Jajuga, Gulce O. Ildiz, and Rui Fausto. 2020. "Thiabendazole and Thiabendazole-Formic Acid Solvate: A Computational, Crystallographic, Spectroscopic and Thermal Study" Molecules 25, no. 13: 3083. https://doi.org/10.3390/molecules25133083
APA StyleTabanez, A. M., Nogueira, B. A., Milani, A., Eusébio, M. E. S., Paixão, J. A., Nur Kabuk, H., Jajuga, M., Ildiz, G. O., & Fausto, R. (2020). Thiabendazole and Thiabendazole-Formic Acid Solvate: A Computational, Crystallographic, Spectroscopic and Thermal Study. Molecules, 25(13), 3083. https://doi.org/10.3390/molecules25133083