
Iron’s Wake: The Performance of Quantum Mechanical-Derived Versus General-Purpose Force Fields Tested on a Luminescent Iron Complex

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
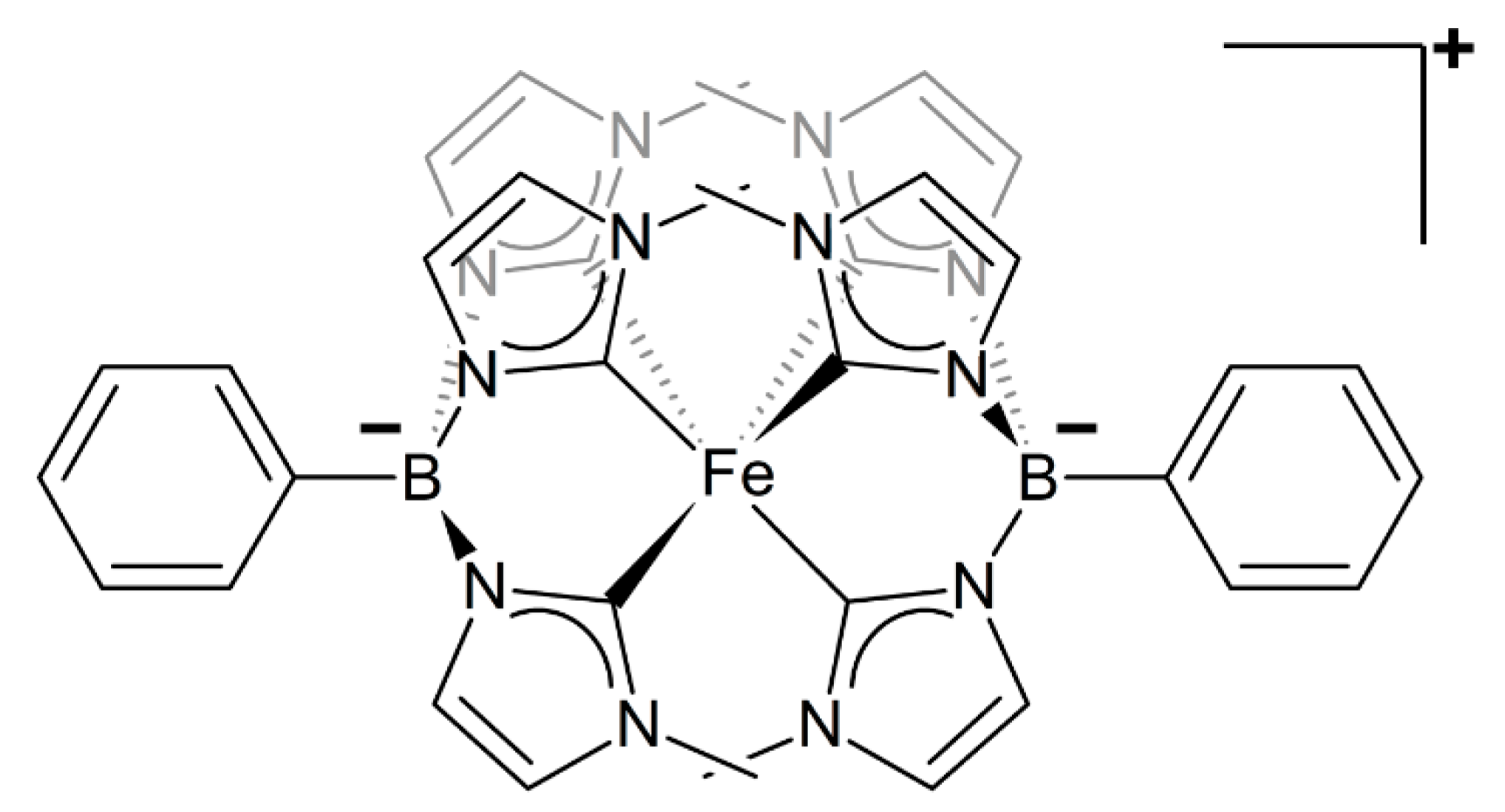
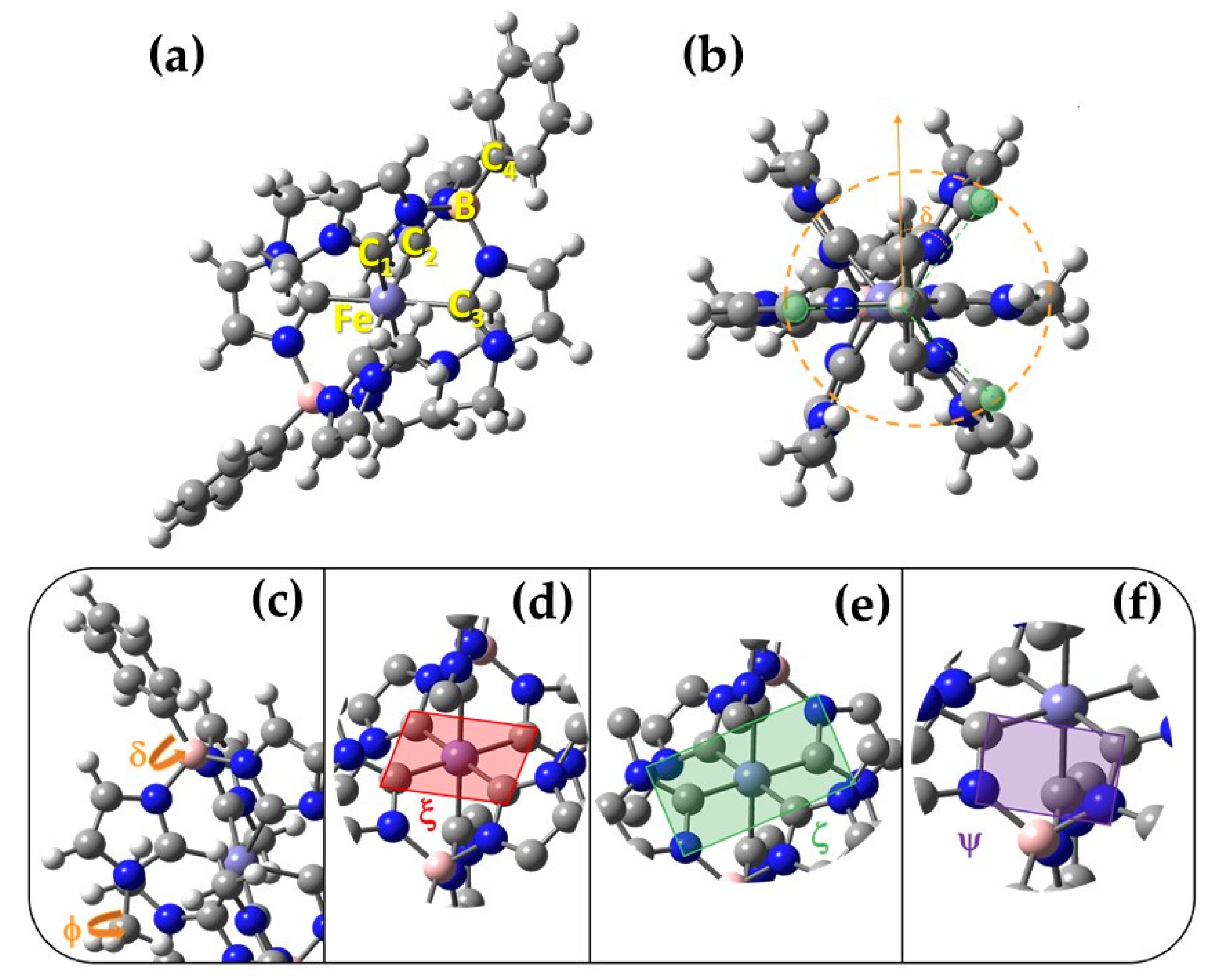
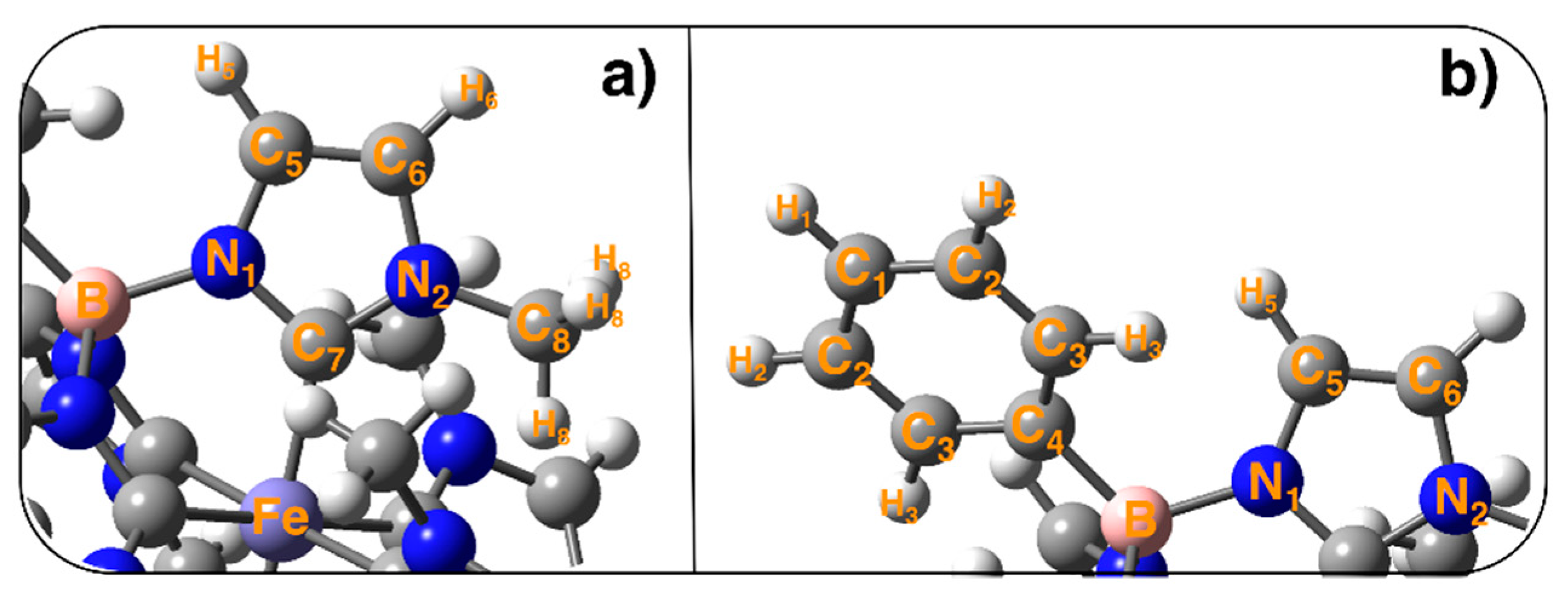
2.1. Structural Properties from MM/MD Simulations
2.1.1. Static Picture: DFT GS Geometry vs Molecular Mechanics
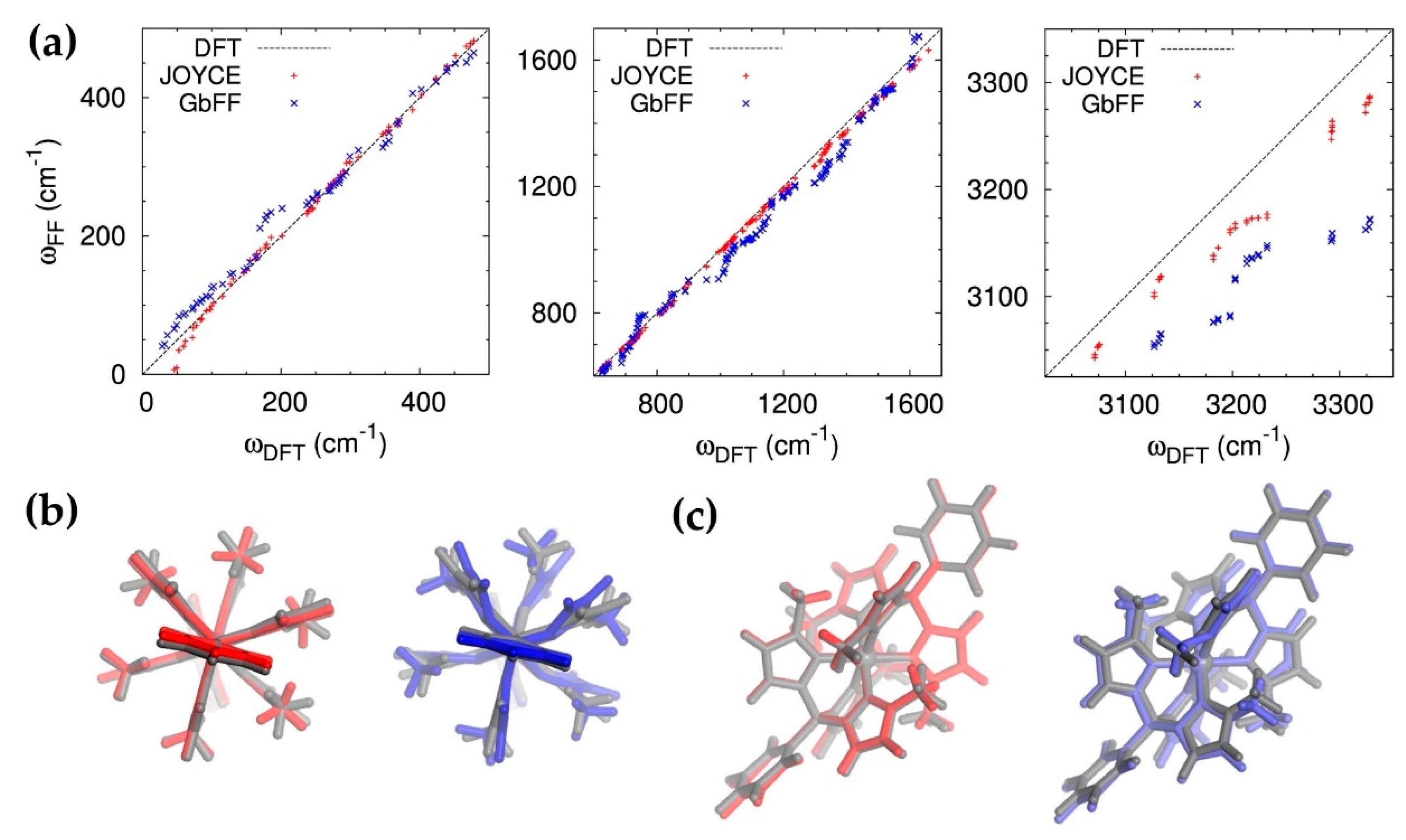
2.1.2. Dynamic Effects and Interaction with the Solvent
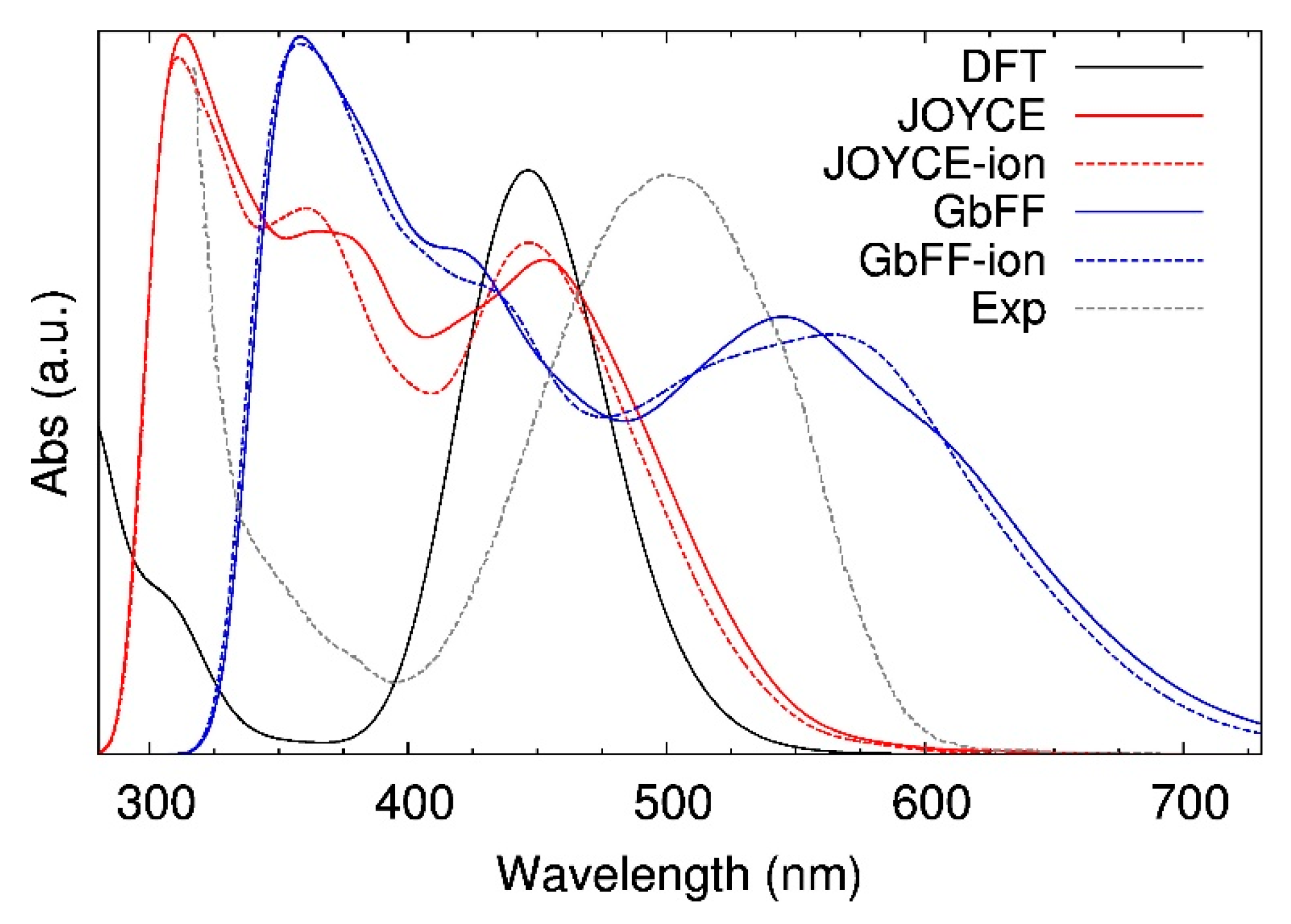
2.2. Optical Properties: Insights from TD-DFT Calculations
2.2.1. Band Assessments from a Static Point of View
2.2.2. Dynamic Averaged Spectra in Solvent
3. Materials and Methods
3.1. Force Field Parametrization
3.1.1. Joyce Force Field
3.1.2. GbFF Parametrization
3.2. MM/MD Simulations
3.3. DFT and TD-DFT Calculations
3.4. Thermally Averaged Absorption Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sension, R.J. Quantum path to photosynthesis. Nature 2007, 446, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Listorti, A.; Durrant, J.; Barber, J. Artificial photosynthesis: Solar to fuel. Nat. Mater. 2009, 8, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Brennaman, M.K.; Dillon, R.J.; Alibabaei, L.; Gish, M.K.; Dares, C.J.; Ashford, D.L.; House, R.L.; Meyer, G.J.; Papanikolas, J.M.; Meyer, T.J. Finding the Way to Solar Fuels with Dye-Sensitized Photoelectrosynthesis Cells. J. Am. Chem. Soc. 2016, 138, 13085–13102. [Google Scholar] [CrossRef] [PubMed]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M.; Nazeeruddin, M.K. Transition Metal Complexes for Photovoltaic and Light Emitting Applications. Struct Bond 2007, 123, 113–175. [Google Scholar]
- Bignozzi, C.A.; Argazzi, R.; Boaretto, R.; Busatto, E.; Carli, S.; Ronconi, F.; Caramori, S. The role of transition metal complexes in dye sensitized solar devices. Coord. Chem. Rev. 2012, 257, 1472–1492. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Wu, W.; Wu, W.; Guo, H.; Sun, J.; Sun, H.; Liu, Y.; Li, Q.; Huang, L. Transition metal complexes with strong absorption of visible light and long-lived triplet excited states: From molecular design to applications. RSC Adv. 2012, 2, 1712–1728. [Google Scholar] [CrossRef]
- Aghazada, S.; Nazeeruddin, M.K. Ruthenium Complexes as Sensitizers in Dye-Sensitized Solar Cells. Inorganics 2018, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Mari, C.; Huang, H.; Rubbiani, R.; Schulze, M.; Würthner, F.; Chao, H.; Gasser, G. Evaluation of Perylene Bisimide-Based Ru II and Ir III Complexes as Photosensitizers for Photodynamic Therapy. Eur. J. Inorg. Chem. 2017, 1745–1752. [Google Scholar] [CrossRef] [Green Version]
- Zamora, A.; Vigueras, G.; Rodríguez, V.; Santana, M.D.; Ruiz, J. Cyclometalated iridium (III) luminescent complexes in therapy and phototherapy. Coord. Chem. Rev. 2018, 360, 34–76. [Google Scholar] [CrossRef]
- Konkankit, C.C.; Marker, S.C.; Knopf, K.M.; Wilson, J.J. Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium. Dalt. Trans. 2018, 47, 9934–9974. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, A.; Van Mourik, F.; Gawelda, W.; Zgrablic, G.; Bressler, C.; Chergui, M. Broadband femtosecond fluorescence spectroscopy of [Ru(bpy) 3]2+. Angew. Chemie Int. Ed. 2006, 45, 3174–3176. [Google Scholar] [CrossRef] [PubMed]
- Chergui, M. Ultrafast Photophysics of Transition Metal Complexes. Acc. Chem. Res. 2015, 48, 801–808. [Google Scholar] [CrossRef]
- Chergui, M. Time-resolved X-ray spectroscopies of chemical systems: New perspectives. Struct. Dyn. 2016, 3, 031001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, P.M.; Zürch, M.; Cushing, S.K.; Neumark, D.M.; Leone, S.R. The ultrafast X-ray spectroscopic revolution in chemical dynamics. Nat. Rev. Chem. 2018, 2, 82–94. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, L.C.; Sarracini, A.; Krawczyk, K.M.; Wentzell, J.S.; Lu, C.; Field, R.L.; Matar, S.F.; Gawelda, W.; Müller-Werkmeister, H.M.; et al. Direct observation of nuclear reorganization driven by ultrafast spin transitions. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Nazeeruddin, M.K.; Klein, C.; Liska, P.; Grätzel, M. Synthesis of novel ruthenium sensitizers and their application in dye-sensitized solar cells. Coord. Chem. Rev. 2005, 249, 1460–1467. [Google Scholar] [CrossRef]
- Han, L.; Islam, A.; Chen, H.; Malapaka, C.; Chiranjeevi, B.; Zhang, S.; Yang, X.; Yanagida, M. High-efficiency dye-sensitized solar cell with a novel co-adsorbent. Energy Environ. Sci. 2012, 5, 6057–6060. [Google Scholar] [CrossRef]
- Pastore, M.; Selloni, A.; Fantacci, S.; De Angelis, F. Electronic and Optical Properties of Dye-Sensitized TiO2 Interfaces. Top Curr. Chem. 2014, 1–45. [Google Scholar] [CrossRef]
- Pastore, M.; De Angelis, F. First-principles modeling of a dye-sensitized TiO2/IrO2 photoanode for water oxidation. J. Am. Chem. Soc. 2015, 137, 5798–5809. [Google Scholar] [CrossRef]
- Arias-Rotondo, D.M.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef] [PubMed]
- Koyyada, G.; Pilli, N.S.; Jung, J.H.; Mandari, K.K.; Shanigaram, B.; Chandrasekharam, M. Shining light on panchromatic ruthenium sensitizers towards dye-sensitized photocatalytic hydrogen evolution. Int. J. Hydrogen Energy 2018, 43, 6963–6976. [Google Scholar] [CrossRef]
- Yun, S.; Qin, Y.; Uhl, A.R.; Vlachopoulos, N.; Yin, M.; Li, D.; Han, X.; Hagfeldt, A. New-generation integrated devices based on dye-sensitized and perovskite solar cells. Energy Environ. Sci. 2018, 11, 476–526. [Google Scholar] [CrossRef]
- McCusker, J.K.; Walda, K.N.; Dunn, R.C.; Simon, J.D.; Magde, D.; Hendrickson, D.N. Subpicosecond 1MLCT → 5T2 Intersystem Crossing of Low-Spin Polypyridyl Ferrous Complexes. J. Am. Chem. Soc. 1993, 115, 298–307. [Google Scholar] [CrossRef]
- Monat, J.E.; McCusker, J.K. Femtosecond excited-state dynamics of an iron(II) polypyridyl solar cell sensitizer model. J. Am. Chem. Soc. 2000, 122, 4092–4097. [Google Scholar] [CrossRef]
- Consani, C.; Prémont-Schwarz, M.; Elnahhas, A.; Bressler, C.; Van Mourik, F.; Cannizzo, A.; Chergui, M. Vibrational coherences and relaxation in the high-spin state of aqueous [FeIIbpy3]2. Angew. Chemie Int. Ed. 2009, 48, 7184–7187. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Gros, P.C.; Assfeld, X.; Monari, A.; Pastore, M. Toward Luminescent Iron Complexes: Unravelling the Photophysics by Computing Potential Energy Surfaces. ChemPhotoChem 2019, 3, 666–683. [Google Scholar] [CrossRef]
- Duchanois, T.; Liu, L.; Pastore, M.; Monari, A.; Cebrián, C.; Trolez, Y.; Darari, M.; Magra, K.; Francés-Monerris, A.; Domenichini, E.; et al. NHC-Based Iron Sensitizers for DSSCs. Inorganics 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Chábera, P.; Kjaer, K.S.; Prakash, O.; Honarfar, A.; Liu, Y.; Fredin, L.A.; Harlang, T.C.B.; Lidin, S.; Uhlig, J.; Sundström, V.; et al. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-To-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Gros, P.C.; Pastore, M.; Assfeld, X.; Monari, A. Photophysical properties of bichromophoric Fe(II) complexes bearing an aromatic electron acceptor. Theor. Chem. Acc. 2019, 138, 1–12. [Google Scholar] [CrossRef]
- Lindh, L.; Chábera, P.; Rosemann, N.W.; Uhlig, J.; Wärnmark, K.; Yartsev, A.; Sundström, V.; Persson, P. Photophysics and photochemistry of iron carbene complexes for solar energy conversion and photocatalysis. Catalysts 2020, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Kaufhold, S.; Wärnmark, K. Design and synthesis of photoactive iron n-heterocyclic carbene complexes. Catalysts 2020, 10, 132. [Google Scholar] [CrossRef] [Green Version]
- Pastore, M.; Duchanois, T.; Liu, L.; Monari, A.; Assfeld, X.; Haacke, S.; Gros, P.C. Interfacial charge separation and photovoltaic efficiency in Fe(II)-carbene sensitized solar cells. Phys. Chem. Chem. Phys. 2016, 18, 28069–28081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Duchanois, T.; Etienne, T.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. A new record excited state 3MLCT lifetime for metalorganic iron(II) complexes. Phys. Chem. Chem. Phys. 2016, 18, 12550–12556. [Google Scholar] [CrossRef] [PubMed]
- Francés-Monerris, A.; Magra, K.; Darari, M.; Cebrián, C.; Beley, M.; Domenichini, E.; Haacke, S.; Pastore, M.; Assfeld, X.; Gros, P.C.; et al. Synthesis and Computational Study of a Pyridylcarbene Fe(II) Complex: Unexpected Effects of fac/mer Isomerism in Metal-to-Ligand Triplet Potential Energy Surfaces. Inorg. Chem. 2018, 57, 10431–10441. [Google Scholar] [CrossRef] [PubMed]
- Magra, K.; Domenichini, E.; Francés-Monerris, A.; Cebrián, C.; Beley, M.; Darari, M.; Pastore, M.; Monari, A.; Assfeld, X.; Haacke, S.; et al. Impact of the fac/mer Isomerism on the Excited-State Dynamics of Pyridyl-carbene Fe(II) Complexes. Inorg. Chem. 2019, 58, 5069–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjær, K.S.; Kaul, N.; Prakash, O.; Chábera, P.; Rosemann, N.W.; Honarfar, A.; Gordivska, O.; Fredin, L.A.; Bergquist, K.E.; Häggström, L.; et al. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. [Google Scholar] [CrossRef]
- Dixon, I.M.; Boissard, G.; Whyte, H.; Alary, F.; Heully, J.L. Computational Estimate of the Photophysical Capabilities of Four Series of Organometallic Iron(II) Complexes. Inorg. Chem. 2016, 55, 5089–5091. [Google Scholar] [CrossRef] [Green Version]
- Wenger, O.S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. [Google Scholar] [CrossRef] [Green Version]
- Wenger, O.S. Is Iron the New Ruthenium? Chem. A Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [Green Version]
- Stock, A. Transition Metals in Coordination Environments; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 29, ISBN 978-3-030-11713-9. [Google Scholar]
- Sousa, C.; Alías, M.; Domingo, A.; de Graaf, C. Deactivation of Excited States in Transition-Metal Complexes: Insight from Computational Chemistry. Chem. A Eur. J. 2019, 25, 1152–1164. [Google Scholar] [CrossRef]
- Amovilli, C.; Barone, V.; Cammi, R.; Cances, E.; Cossi, M.; Menucci, B.; Pomelli, C.S.; Tomasi, J. Recent advances in the description of solvent effects with the polarizable continuum model. Adv. Quantum Chem. 1999, 32, 227–261. [Google Scholar]
- Hush, N.S.; Reimers, J.R. Solvent Effects on the Electronic Spectra of Transition Metal Complexes. Chem. Rev. 2000, 100, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Véry, T.; Ambrosek, D.; Otsuka, M.; Gourlaouen, C.; Assfeld, X.; Monari, A.; Daniel, C. Photophysical properties of ruthenium(ii) polypyridyl dna intercalators: Effects of the molecular surroundings investigated by theory. Chem. A Eur. J. 2014, 20, 12901–12909. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Meyer, T.J. Medium effects on charge transfer in metal complexes. Chem. Rev. 1998, 98, 1439–1477. [Google Scholar] [CrossRef]
- Moret, M.E.; Tavernelli, I.; Chergui, M.; Rothlisberger, U. Electron localization dynamics in the triplet excited state of [Ru(bpy)3]2+ in aqueous solution. Chem. A Eur. J. 2010, 16, 5889–5894. [Google Scholar] [CrossRef]
- Rondi, A.; Rodriguez, Y.; Feurer, T.; Cannizzo, A. Solvation-driven charge transfer and localization in metal complexes. Acc. Chem. Res. 2015, 48, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Kjær, K.S.; Zhang, W.; Alonso-Mori, R.; Bergmann, U.; Chollet, M.; Hadt, R.G.; Hartsock, R.W.; Harlang, T.; Kroll, T.; Kubiček, K.; et al. Ligand manipulation of charge transfer excited state relaxation and spin crossover in [Fe(2,2′-bipyridine)2(CN)2]. Struct. Dyn. 2017, 4, 044030. [Google Scholar] [CrossRef] [Green Version]
- Pápai, M.; Abedi, M.; Levi, G.; Biasin, E.; Nielsen, M.M.; Møller, K.B. Theoretical Evidence of Solvent-Mediated Excited-State Dynamics in a Functionalized Iron Sensitizer. J. Phys. Chem. C 2019, 123, 2056–2065. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, S.; Rocca, D.; Pastore, M. Role of Solvent in the Energy Level Alignment of Dye-Sensitized NiO Interfaces. J. Phys. Chem. C 2017, 121, 22286–22294. [Google Scholar] [CrossRef]
- Shao, Y.; De Ruiter, J.M.; De Groot, H.J.M.; Buda, F. Photocatalytic Water Splitting Cycle in a Dye-Catalyst Supramolecular Complex: Ab Initio Molecular Dynamics Simulations. J. Phys. Chem. C 2019, 123, 21403–21414. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.-Y.; Jia, J.; Santra, B.; Wu, X.; Car, R.; DiStasio, R.A., Jr. Enabling Large-Scale Condensed-Phase Hybrid Density Functional Theory Based Ab Initio Molecular Dynamics 1: Theory, Algorithm, and Performance. J. Chem. Theory Comput. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monari, A.; Rivail, J.-L.; Assfeld, X. Theoretical Modeling of Large Molecular Systems. Advances in the Local Self Consistent Field Method for Mixed Quantum Mechanics/Molecular Mechanics Calculations. Acc. Chem. Res. 2013, 46, 596–603. [Google Scholar] [CrossRef]
- Morzan, U.N.; Alonso De Armiño, D.J.; Foglia, N.O.; Ramírez, F.; González Lebrero, M.C.; Scherlis, D.A.; Estrin, D.A. Spectroscopy in Complex Environments from QM-MM Simulations. Chem. Rev. 2018, 118, 4071–4113. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Schreiner, P.R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chemie Int. Ed. 2018, 57, 4170–4176. [Google Scholar] [CrossRef]
- Prampolini, G.; Ingrosso, F.; Cerezo, J.; Iagatti, A.; Foggi, P.; Pastore, M. Short- and Long-Range Solvation Effects on the Transient UV-Vis Absorption Spectra of a Ru(II)-Polypyridine Complex Disentangled by Nonequilibrium Molecular Dynamics. J. Phys. Chem. Lett. 2019, 10, 2885–2891. [Google Scholar] [CrossRef]
- Jahn, D.A.; Akinkunmi, F.O.; Giovambattista, N. Effects of temperature on the properties of glycerol: A computer simulation study of five different force fields. J. Phys. Chem. B 2014, 118, 11284–11294. [Google Scholar] [CrossRef]
- Prampolini, G.; Livotto, P.R.; Cacelli, I. Accuracy of quantum mechanically derived force-fields parameterized from dispersion-corrected DFT data: The benzene dimer as a prototype for aromatic interactions. J. Chem. Theory Comput. 2015, 11, 5182–5196. [Google Scholar] [CrossRef]
- Cerezo, J.; Santoro, F.; Prampolini, G. Comparing classical approaches with empirical or quantum-mechanically derived force fields for the simulation electronic lineshapes: Application to coumarin dyes. Theor. Chem. Acc. 2016, 135, 1–21. [Google Scholar] [CrossRef]
- Andreussi, O.; Prandi, I.G.; Campetella, M.; Prampolini, G.; Mennucci, B. Classical Force Fields Tailored for QM Applications: Is It Really a Feasible Strategy? J. Chem. Theory Comput. 2017, 13, 4636–4648. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Rappé, A.K.; Colwell, K.S.; Casewit, C.J. Application of a Universal Force Field to Metal Complexes. Inorg. Chem. 1993, 32, 3438–3450. [Google Scholar] [CrossRef]
- Lin, F.; Wang, R. Systematic derivation of AMBER force field parameters applicable to zinc-containing systems. J. Chem. Theory Comput. 2010, 6, 1852–1870. [Google Scholar] [CrossRef]
- Hu, L.; Ryde, U. Comparison of methods to obtain force-field parameters for metal sites. J. Chem. Theory Comput. 2011, 7, 2452–2463. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.Y.; Ponder, J.W. An angular overlap model for Cu(II) ion in the AMOEBA polarizable force field. J. Chem. Theory Comput. 2014, 10, 298–311. [Google Scholar] [CrossRef] [Green Version]
- Vaiana, A.C.; Schulz, A.; Wolfrum, J.; Sauer, M.; Smith, J.C. Molecular mechanics force field parameterization of the fluorescent probe rhodamine 6G using automated frequency matching. J. Comput. Chem. 2003, 24, 632–639. [Google Scholar] [CrossRef]
- Nilsson, K.; Lecerof, D.; Sigfridsson, E.; Ryde, U. An automatic method to generate force-field parameters for hetero-compounds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 274–289. [Google Scholar] [CrossRef]
- Allen, A.E.A.; Payne, M.C.; Cole, D.J. Harmonic Force Constants for Molecular Mechanics Force Fields via Hessian Matrix Projection. J. Chem. Theory Comput. 2018, 14, 274–281. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo Santos, L.; Prandi, I.G.; Ramalho, T.C. Could Quantum Mechanical Properties Be Reflected on Classical Molecular Dynamics? The Case of Halogenated Organic Compounds of Biological Interest. Front. Chem. 2019, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.T.; Allen, A.E.A.; Dodda, L.S.; Cole, D.J. QUBEKit: Automating the Derivation of Force Field Parameters from Quantum Mechanics. J. Chem. Inf. Model. 2019, 59, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Prampolini, G.; Ingrosso, F.; Segalina, A.; Caramori, S.; Foggi, P.; Pastore, M. Dynamical and Environmental Effects on the Optical Properties of an Heteroleptic Ru(II)-Polypyridine Complex: A Multilevel Approach Combining Accurate Ground and Excited State QM-Derived Force Fields, MD and TD-DFT. J. Chem. Theory Comput. 2019, 15, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. A general quantum mechanically derived force field (QMDFF) for molecules and condensed phase simulations. J. Chem. Theory Comput. 2014, 10, 4497–4514. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; De Mitri, N.; Licari, D.; Monti, S.; Prampolini, G. Joyce and Ulysses: Integrated and user-friendly tools for the parameterization of intramolecular force fields from quantum mechanical data. Phys. Chem. Chem. Phys. 2013, 15, 3736–3751. [Google Scholar] [CrossRef]
- Piquemal, J.P.; Jordan, K.D. From quantum mechanics to force fields: New methodologies for the classical simulation of complex systems. Theor. Chem. Acc. 2012, 131, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Waldher, B.; Kuta, J.; Chen, S.; Henson, N.; Clark, A.E. ForceFit: A Code to Fit Classical Force Fields to Quantum Mechanical Potential Energy Surfaces. J. Comput. Chem. 2010, 31, 2307–2316. [Google Scholar] [CrossRef]
- Akin-Ojo, O.; Song, Y.; Wang, F. Developing ab initio quality force fields from condensed phase quantum-mechanics/molecular-mechanics calculations through the adaptive force matching method. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef]
- Verstraelen, T.; Van Neck, D.; Ayers, P.W.; Van Speybroeck, V.; Waroquier, M. The gradient curves method: An improved strategy for the derivation of molecular mechanics valence force fields from ab initio data. J. Chem. Theory Comput. 2007, 3, 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Vaiana, A.C.; Ullmann, G.M.; Smith, J.C. Derivation of a molecular mechanics force field for cholesterol. Pure Appl. Chem. 2004, 76, 189–196. [Google Scholar] [CrossRef]
- Rosemann, N.W.; Chábera, P.; Prakash, O.; Kaufhold, S.; Wärnmark, K.; Yartsev, A.; Persson, P. Tracing the Full Bimolecular Photocycle of Iron(III)–Carbene Light Harvesters in Electron-Donating Solvents. J. Am. Chem. Soc. 2020, 142, 8565–8569. [Google Scholar] [CrossRef] [PubMed]
- Cacelli, I.; Prampolini, G. Parametrization and validation of intramolecular force fields derived from DFT calculations. J. Chem. Theory Comput. 2007, 3, 1803–1817. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, J.; Prampolini, G.; Cacelli, I. Developing accurate intramolecular force fields for conjugated systems through explicit coupling terms. Theor. Chem. Acc. 2018, 137, 1–15. [Google Scholar] [CrossRef]
- Prampolini, G.; Yu, P.; Pizzanelli, S.; Cacelli, I.; Yang, F.; Zhao, J.; Wang, J. Structure and dynamics of ferrocyanide and ferricyanide anions in water and heavy water: An insight by MD simulations and 2D IR spectroscopy. J. Phys. Chem. B 2014, 118, 14899–14912. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 56531, 1157–1174. [Google Scholar] [CrossRef]
- Cacelli, I.; Prampolini, G. Torsional barriers and correlations between dihedrals in p-polyphenyls. J. Phys. Chem. A 2003, 107, 8665–8670. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Price, M.L.P.; Ostrovsky, D.; Jorgensen, W.L. Gas-phase and liquid-state properties of esters, nitriles, and nitro compounds with the OPLS-AA force field. J. Comput. Chem. 2001, 22, 1340–1352. [Google Scholar] [CrossRef]
- Sambasivarao, S.V.; Acevedo, O. Development of OPLS-AA force field parameters for 68 unique ionic liquids. J. Chem. Theory Comput. 2009, 5, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A., III. DREIDING: A Generic Force Field for Molecular Simulations. J. Phys. Chem. 1990, 101, 8897–8909. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. MCPB. py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Gattuso, H.; Duchanois, T.; Besancenot, V.; Barbieux, C.; Assfeld, X.; Becuwe, P.; Gros, P.C.; Grandemange, S.; Monari, A. Interaction of Iron II Complexes with B-DNA. Insights from Molecular Modeling, Spectroscopy, and Cellular Biology. Front. Chem. 2015, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tafi, A.; Agamennone, M.; Tortorella, P.; Alcaro, S.; Gallina, C.; Botta, M. AMBER force field implementation of the boronate function to simulate the inhibition of b-lactamases by alkyl and aryl boronic acids. Eur. J. Med. Chem. 2005, 40, 1134–1142. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M. Parameterization of highly charged metal ions using the 12-6-4 LJ-type nonbonded model in explicit water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01 2016; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Nikitin, A.M.; Lyubartsev, A.P. New Six-site Acetonitrile Model for Simulations of Liquid Acetonitrile and its Aqueous Mixtures. J. Comput. Chem. 2007, 12, 2020–2026. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, S.; Wang, W. A Refined Force Field for Molecular Simulation of Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2004, 108, 12978–12989. [Google Scholar] [CrossRef]
- Sousa, A.W.; Vranken, W.F. Open Access ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Fredin, L.A.; Persson, P. Exceptional Excited-State Lifetime of an Iron(II)–N-Heterocyclic Carbene Complex Explained. J. Phys. Chem. Lett. 2014, 5, 2066–2071. [Google Scholar] [CrossRef]
- Dixon, I.M.; Alary, F.; Boggio-Pasqua, M.; Heully, J. Reversing the relative 3MLCT–3MC order in Fe(II) complexes using cyclometallating ligands: A computational study aiming at luminescent Fe(II) complexes. Dalt. Trans. 2015, 44, 13498–13503. [Google Scholar] [CrossRef]
- Kepp, K.P. Theoretical Study of Spin Crossover in 30 Iron Complexes. Inorg. Chem. 2016, 55, 2717–2727. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | DFT | Joyce | GbFF | XRD [PF6] 1 | XRD [BPh4] 1 | |
|---|---|---|---|---|---|---|
| Fe-C1 | 2.041 | 2.040 | 2.055 | 2.008 | 1.984 | |
| Bond distances (Å) | Fe-C2 | 2.027 | 2.026 | 2.052 | 2.002 | 1.979 |
| Fe-C3 | 1.998 | 1.999 | 2.048 | 1.979 | 1.971 | |
| Fe-B | 3.215 | 3.215 | 3.421 | 3.202 | 3.182 | |
| Bond angles (°) | C1-Fe-C2 | 85.57 | 85.64 | 82.31 | 86.49 | 86.47 |
| C1-Fe-C3 | 86.77 | 86.67 | 83.36 | 87.00 | 86.86 | |
| C2-Fe-C3 | 87.47 | 87.53 | 84.52 | 87.24 | 86.86 | |
| Fe-B-C4 | 173.4 | 173.4 | 175.5 | 173.7 | 174.4 |
| FF Method | Bond Length (Å) | Bending Angle (°) | Dihedrals (°) |
|---|---|---|---|
| Joyce | 0.00 | 0.07 | 15.89 |
| GbFF | 0.03 | 2.75 | 22.13 |
| Method | State | λ(nm) | f | Fragment | h+ | e− |
|---|---|---|---|---|---|---|
| Fe | 0.007 | 0.328 | ||||
| CL1 | 0.306 | 0.188 | ||||
| DFT | D4 | 451 (2.75) | 0.071 | CL2 | 0.266 | 0.3 |
| CL3 | 0.414 | 0.177 | ||||
| ΣCL | 0.985 | 0.666 | ||||
| Fe | 0.008 | 0.332 | ||||
| CL1 | 0.306 | 0.193 | ||||
| Joyce | D4 | 449 (2.76) | 0.073 | CL2 | 0.263 | 0.292 |
| CL3 | 0.414 | 0.176 | ||||
| ΣCL | 0.984 | 0.661 | ||||
| Fe | 0.018 | 0.31 | ||||
| CL1 | 0.454 | 0.169 | ||||
| GbFF | D7 | 531 (2.34) | 0.048 | CL2 | 0.24 | 0.246 |
| CL3 | 0.271 | 0.253 | ||||
| ΣCL | 0.965 | 0.668 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diez-Cabanes, V.; Prampolini, G.; Francés-Monerris, A.; Monari, A.; Pastore, M. Iron’s Wake: The Performance of Quantum Mechanical-Derived Versus General-Purpose Force Fields Tested on a Luminescent Iron Complex. Molecules 2020, 25, 3084. https://doi.org/10.3390/molecules25133084
Diez-Cabanes V, Prampolini G, Francés-Monerris A, Monari A, Pastore M. Iron’s Wake: The Performance of Quantum Mechanical-Derived Versus General-Purpose Force Fields Tested on a Luminescent Iron Complex. Molecules. 2020; 25(13):3084. https://doi.org/10.3390/molecules25133084
Chicago/Turabian StyleDiez-Cabanes, Valentin, Giacomo Prampolini, Antonio Francés-Monerris, Antonio Monari, and Mariachiara Pastore. 2020. "Iron’s Wake: The Performance of Quantum Mechanical-Derived Versus General-Purpose Force Fields Tested on a Luminescent Iron Complex" Molecules 25, no. 13: 3084. https://doi.org/10.3390/molecules25133084
APA StyleDiez-Cabanes, V., Prampolini, G., Francés-Monerris, A., Monari, A., & Pastore, M. (2020). Iron’s Wake: The Performance of Quantum Mechanical-Derived Versus General-Purpose Force Fields Tested on a Luminescent Iron Complex. Molecules, 25(13), 3084. https://doi.org/10.3390/molecules25133084