
Determination of Polypeptide Antibiotic Residues in Food of Animal Origin by Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry


Abstract
:1. Introduction
2. Results and Discussion
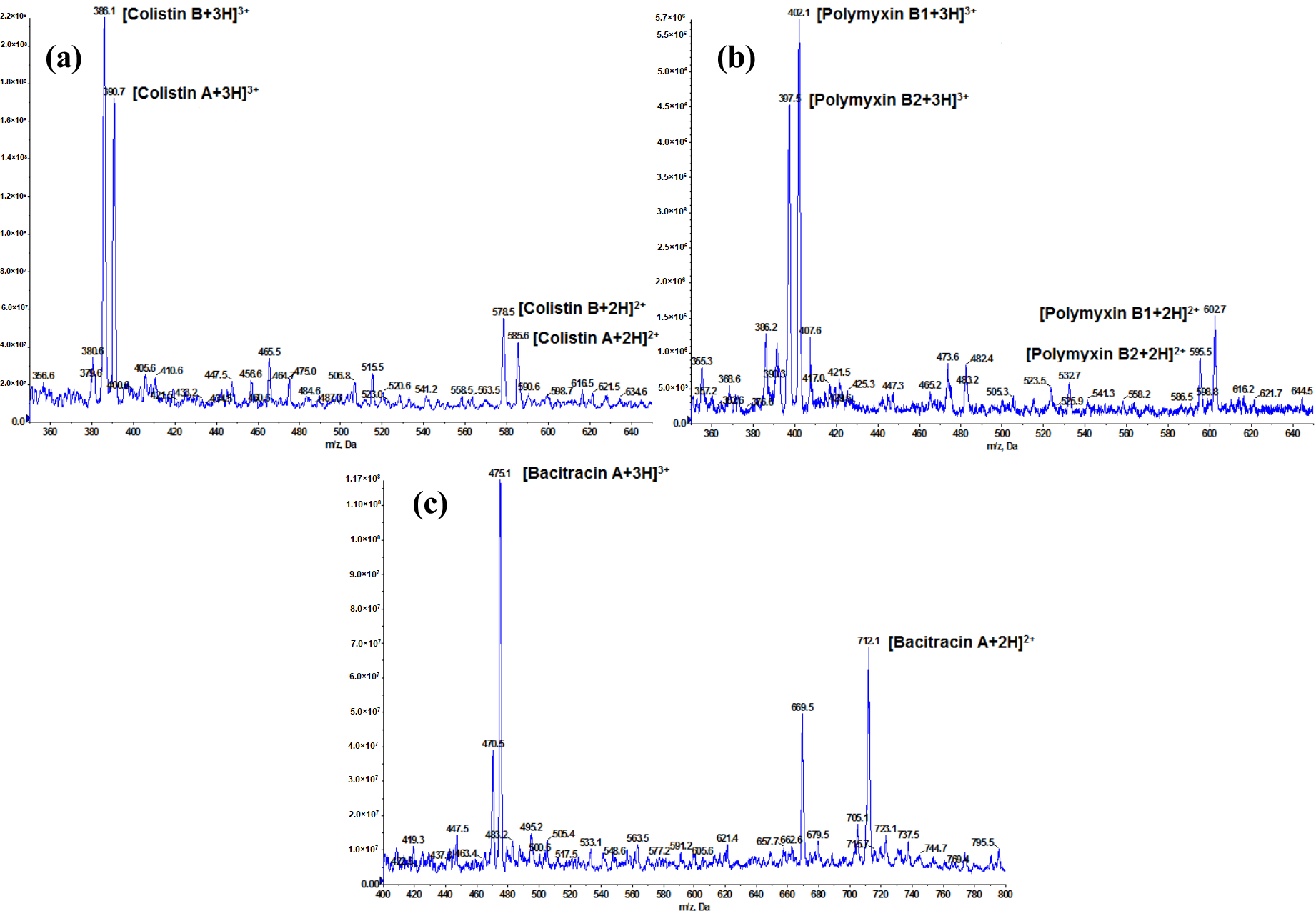
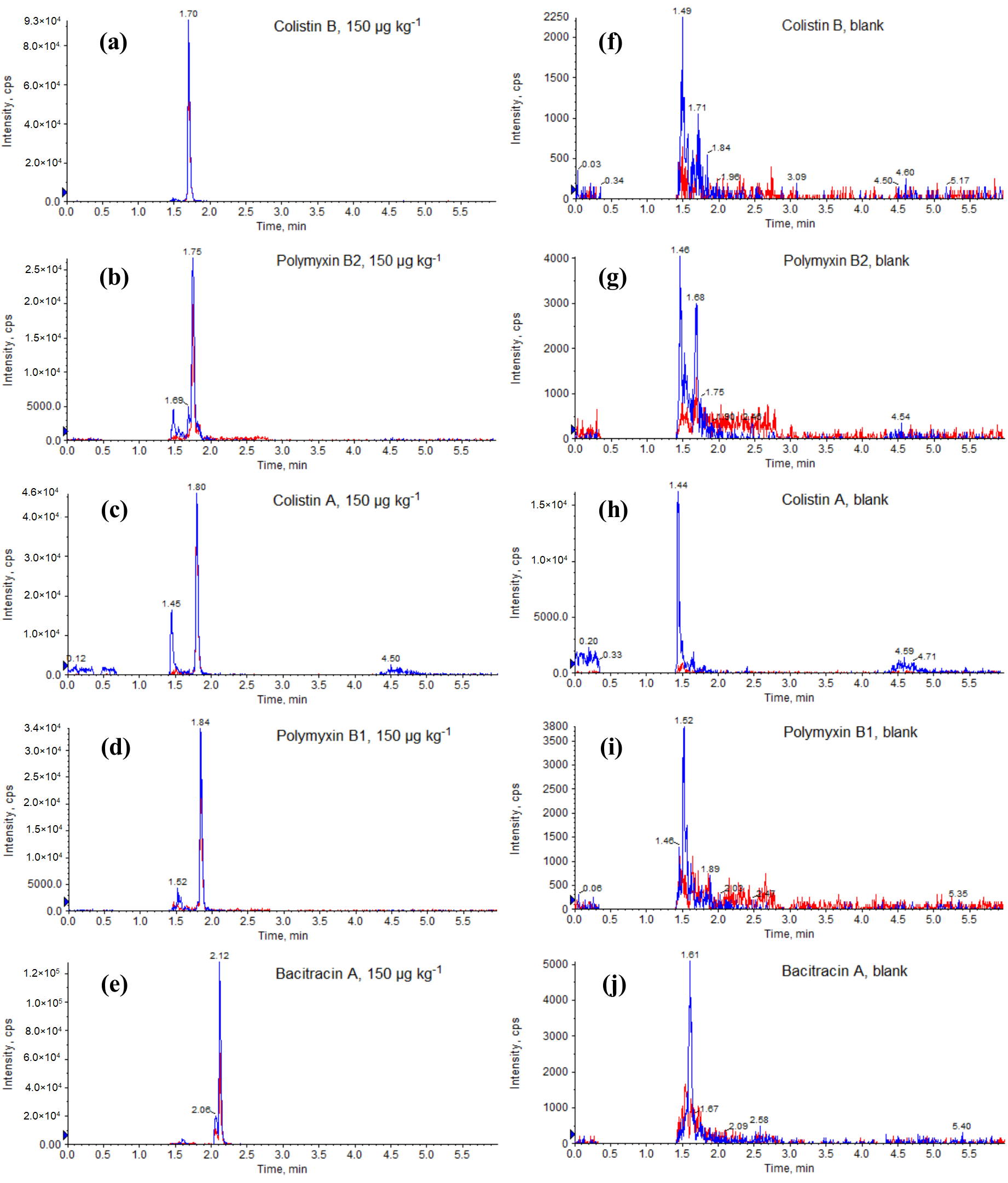
2.1. Optimization of LC-MS/MS Conditions
2.2. Optimization of Sample Preparation
2.3. Method Validation
3. Materials and Methods
3.1. Chemical and Reagents
3.2. LC-MS/MS Conditions
3.3. Sample Preparation
3.4. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- The European Agency for the Evaluation of Medical Products. Committee for Medicinal Products for Veterinary Use. Bacitracin. Summary Report (2). EMEA/MRL/768/00-FINAL, January 2001. Available online: https://www.ema.europa.eu/en/documents/mrl-report/bacitracin-summary-report-2-committee-veterinary-medicinal-products_en.pdf (accessed on 3 March 2020).
- Ma, Z.; Wang, J.; Gerber, J.P.; Milne, R.W. Determination of colistin in human plasma, urine and other biological samples using LC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 862, 205–212. [Google Scholar] [CrossRef]
- Covelli, J.; Ruszaj, D.; Straubinger, R.; Li, J.; Rao, G.G. The development and validation of a simple liquid chromatography-tandem mass spectrometry method for polymyxin B1 and B2 quantification in different matrices. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1065, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Catry, B.; Cavaleri, M.; Baptiste, K.; Grave, K.; Grein, K.; Holm, A.; Jukes, H.; Liebana, E.; Lopez Navas, A.; Mackay, D.; et al. Use of colistin-containing products within the European Union and European Economic Area (EU/EEA): Development of resistance in animals and possible impact on human and animal health. Int. J. Antimicrob. Agents 2015, 46, 297–306. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency, European Surveillance of Veterinary Antimicrobial Consumption, 2018. Sales of Veterinary Antimicrobial Agents in 30 European Countries in 2016. (EMA/275982/2018). Available online: https://www.ema.europa.eu/en/documents/report/sales-veterinary-antimicrobial-agents-30-european-countries-2016-trends-2010-2016-eighth-esvac_en.pdf (accessed on 3 March 2020).
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- European Communities. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Communities 2010, L15, 1–72. [Google Scholar]
- Sin, D.W.M.; Ho, C.; Wong, Y.C.; Ho, S.K.; Ip, A.C.B. Analysis of major components of residual bacitracin and colistin in food samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2005, 535, 23–31. [Google Scholar] [CrossRef]
- Wan, E.C.H.; Ho, C.; Sin, D.W.M.; Wong, Y.C. Detection of residual bacitracin A, colistin A, and colistin B in milk and animal tissues by liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2006, 385, 181–188. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, X.; Ren, C.; Huang, H.; Zhang, X.; Gong, X.; Liu, H.; Yu, Z.; Zhang, L. Analysis of colistin A and B in fishery products by ultra performance liquid chromatography with positive electrospray ionization tandem mass spectrometry. J. Chromatogr. B 2012, 899, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Park, J.A.; Kim, D.S.; Kim, N.H.; Kim, S.K.; Cho, K.S.; Jeong, D.; Shim, J.H.; El-Aty, A.M.A.; Shin, H.C. Simultaneous detection of bacitracin and polymyxin B in livestock products using liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2015, 38, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Li, X.; Zheng, K.; Ke, Y.; Wang, Y.; Wang, L.; Yu, F.; Xia, X. Determination of colistin in animal tissues, egg, milk, and feed by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 248, 166–172. [Google Scholar] [CrossRef]
- Saluti, G.; Diamanti, I.; Giusepponi, D.; Pucciarini, L.; Rossi, R.; Moretti, S.; Sardella, R.; Galarini, R. Simultaneous determination of aminoglycosides and colistins in food. Food Chem. 2018, 266, 9–16. [Google Scholar] [CrossRef]
- Kaufmann, A.; Widmer, M. Quantitative analysis of polypeptide antibiotic residues in a variety of food matrices by liquid chromatography coupled to tandem mass spectrometry. Anal. Chim. Acta 2013, 797, 81–88. [Google Scholar] [CrossRef]
- Boison, J.O.; Lee, S.; Matus, J. A multi-residue method for the determination of seven polypeptide drug residues in chicken muscle tissues by LC-MS/MS. Anal. Bioanal. Chem. 2015, 407, 4065–4078. [Google Scholar] [CrossRef]
- European Communities. Commission Decision (2002/657/EC) of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, L221, 8–36. [Google Scholar]
- Rauh, M. LC-MS/MS for protein and peptide quantification in clinical chemistry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 883, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.L.; Le Blanc, J.C. Peptide and protein drug analysis by MS: Challenges and opportunities for the discovery environment. Bioanalysis 2011, 3, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, S.A.; Song, F.; Su, M.; Hang, T.; Song, M. Analysis of bacitracin and its related substances by liquid chromatography tandem mass spectrometry. J. Pharm. Anal. 2017, 7, 48–55. [Google Scholar] [CrossRef]
- Govaerts, C.; Rozenski, J.; Orwa, J.; Roets, E.; Van Schepdael, A.; Hoogmartens, J. Mass spectrometric fragmentation of cyclic peptides belonging to the polymyxin and colistin antibiotics studied by ion trap and quadrupole/orthogonal-acceleration time-of-flight technology. Rapid Commun. Mass Spectrom. 2002, 16, 823–833. [Google Scholar] [CrossRef]
- Govaerts, C.; Li, C.; Orwa, J.; Van Schepdael, A.; Adams, E.; Roets, E.; Hoogmartens, J. Sequencing of bacitracin A and related minor components by liquid chromatography/electrospray ionization ion trap tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 1366–1379. [Google Scholar] [CrossRef]
- Berendsen, B.J.; Stolker, L.A.; Nielen, M.W. Selectivity in the sample preparation for the analysis of drug residues in products of animal origin using LC-MS. Trends Anal. Chem. 2013, 43, 229–239. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Development of an improved high resolution mass spectrometry based multi-residue method for veterinary drugs in various food matrices. Anal. Chim. Acta. 2011, 700, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.A.; Vera, J.H. Effect of acids and bases on the solubility of amino acids. Fluid Phase Equilibr. 1998, 152, 121–132. [Google Scholar] [CrossRef]
- Tseng, H.C.; Lee, C.Y.; Wen-Lu Weng, W.L.; Shiah, I.M. Solubilities of amino acids in water at various pH values under 298.15K. Fluid Phase Equilibr. 2009, 285, 90–95. [Google Scholar] [CrossRef]
- Gekko, K.; Ohmae, E.; Kameyama, K.; Takagi, T. Acetonitrile-protein interactions: Amino acid solubility and preferential solvation. Biochim. Biophys. Acta. 1998, 1387, 195–205. [Google Scholar] [CrossRef]
- Bowden, N.A.; Sanders, J.P.M.; Bruins, M.E. Solubility of the Proteinogenic α-Amino Acids in Water, Ethanol, and Ethanol-Water Mixtures. J. Chem. Eng. Data 2018, 63, 488–497. [Google Scholar] [CrossRef]
- Chemicalize. Available online: https://chemicalize.com (accessed on 3 March 2020).
- Dotsikas, Y.; Markopoulou, C.K.; Koundourellis, J.E.; Loukas, Y.L. Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J. Sep. Sci. 2011, 34, 37–45. [Google Scholar] [CrossRef]
- SANCO/2004/2726-rev 4-December 2008. Guidelines for the implementation of Decision 2002/657/EC. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_vet-med-residues_cons_2004-2726rev4_en.pdf (accessed on 10 July 2020).
- Song, X.; Huang, Q.; Zhang, Y.; Zhang, M.; Xie, J.; He, L. Rapid multiresidue analysis of authorized/banned cyclopolypeptide antibiotics in feed by liquid chromatography-tandem mass spectrometry based on dispersive solid-phase extraction. J. Pharm. Biomed. Anal. 2019, 170, 234–242. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Bacitracin A | Colistin A | Colistin B | Polymyxin B1 | Polymyxin B2 |
|---|---|---|---|---|---|
| Spiked level (µg kg−1) | 10/75/150/225 | 10/75/150/225 | 10/75/150/225 | 10/75/150/225 | 10/75/150/225 |
| Recovery (%) | 91/93/95/95 | 97/95/94/96 | 94/93/94/97 | 99/93/95/95 | 97/93/94/96 |
| Repeatability (CV, %) | 9.8/7.2/6.8/6.3 | 9.7/7.9/7.8/7.4 | 9.0/7.3/6.9/6.0 | 9.9/9.7/8.8/8.5 | 9.8/9.3/8.6/7.7 |
| Within-lab reproducibility (CV, %) | 10.4/9.0/8.6/7.7 | 12.6/10.4/10.5/9.4 | 10.4/10.1/9.5/8.4 | 12.0/11.0/9.9/9.7 | 12.2/10.2/9.6/8.8 |
| CCα (µg kg−1) | 168 | 172 | 177 | 14.3 | 13.6 |
| CCβ (µg kg−1) | 192 | 203 | 206 | 17.8 | 16.7 |
| Matrix effect (%) | −62 | −49 | −48 | −66 | −65 |
| Parameter | Bacitracin A | Colistin A | Colistin B | Polymyxin B1 | Polymyxin B2 |
|---|---|---|---|---|---|
| Spiked level (µg kg−1) | 10/50/100/150 | 10/25/50/75 | 10/25/50/75 | 10/25/50/75 | 10/25/50/75 |
| Recovery (%) | 96/98/98/96 | 89/84/83/85 | 86/82/79/78 | 79/78/78/81 | 80/81/80/83 |
| Repeatability (CV, %) | 11.1/9.3/9.0/8.9 | 11.6/10.2/9.8/8.9 | 11.3/10.6/10.5/9.7 | 12.4/12.2/10.4/9.7 | 12.7/10.8/10.0/9.5 |
| Within-lab reproducibility (CV, %) | 13.4/10.0/10.2/9.6 | 13.7/12.3/12.0/10.5 | 13.3/11.2/11.0/10.4 | 13.9/13.1/11.6/10.5 | 13.4/12.0/10.6/10.3 |
| CCα (µg kg−1) | 117 | 60.5 | 59.0 | 13.1 | 13.1 |
| CCβ (µg kg−1) | 140 | 73.7 | 71.8 | 16.6 | 16.5 |
| Matrix effect (%) | −65 | −35 | −33 | −38 | −41 |
| Parameter | Bacitracin A | Colistin A | Colistin B | Polymyxin B1 | Polymyxin B2 |
|---|---|---|---|---|---|
| Spiked level (µg kg−1) | 10/150/300/450 | 10/150/300/450 | 10/150/300/450 | 10/150/300/450 | 10/150/300/450 |
| Recovery (%) | 83/80/83/83 | 71/74/72/73 | 71/75/75/74 | 70/75/74/73 | 72/74/72/72 |
| Repeatability (CV, %) | 10.4/9.7/8.2/7.0 | 10.8/7.7/7.3/7.2 | 11.5/7.1/6.9/6.5 | 10.5/8.1/7.9/7.3 | 9.2/7.2/6.9/6.8 |
| Within-lab reproducibility (CV, %) | 11.2/11.5/9.6/8.8 | 12.1/10.5/9.8/9.6 | 13.2/10.7/9.6/8.8 | 12.0/11.5/10.3/9.7 | 11.2/10.1/8.7/8.7 |
| CCα (µg kg−1) | 13.9 | 340 | 343 | 14.3 | 14.1 |
| CCβ (µg kg−1) | 18.4 | 402 | 400 | 18.3 | 17.9 |
| Matrix effect (%) | −25 | −28 | −26 | −29 | −34 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bladek, T.; Szymanek-Bany, I.; Posyniak, A. Determination of Polypeptide Antibiotic Residues in Food of Animal Origin by Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2020, 25, 3261. https://doi.org/10.3390/molecules25143261
Bladek T, Szymanek-Bany I, Posyniak A. Determination of Polypeptide Antibiotic Residues in Food of Animal Origin by Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Molecules. 2020; 25(14):3261. https://doi.org/10.3390/molecules25143261
Chicago/Turabian StyleBladek, Tomasz, Iwona Szymanek-Bany, and Andrzej Posyniak. 2020. "Determination of Polypeptide Antibiotic Residues in Food of Animal Origin by Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry" Molecules 25, no. 14: 3261. https://doi.org/10.3390/molecules25143261
APA StyleBladek, T., Szymanek-Bany, I., & Posyniak, A. (2020). Determination of Polypeptide Antibiotic Residues in Food of Animal Origin by Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Molecules, 25(14), 3261. https://doi.org/10.3390/molecules25143261