
Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators



Abstract
:1. Introduction
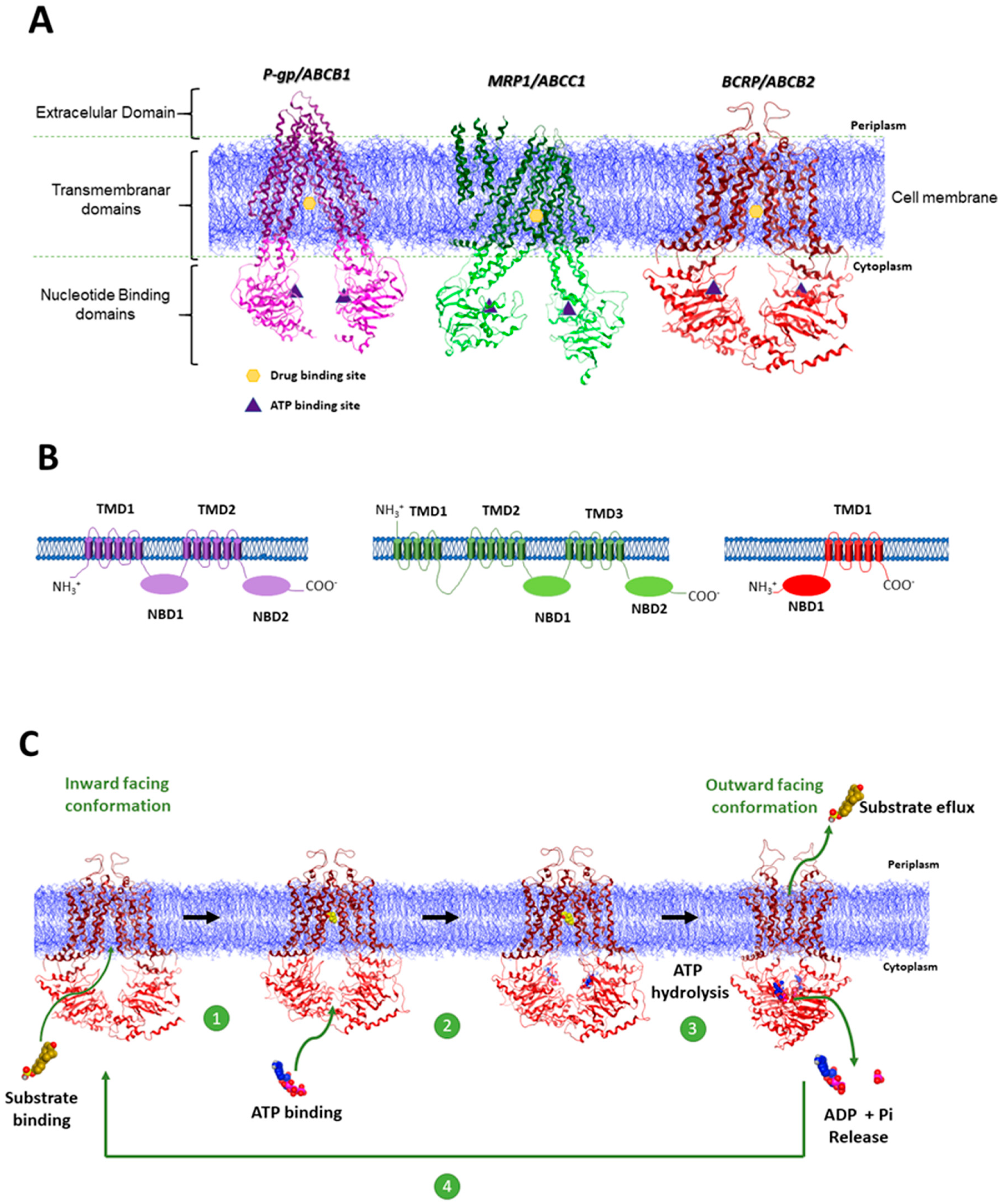
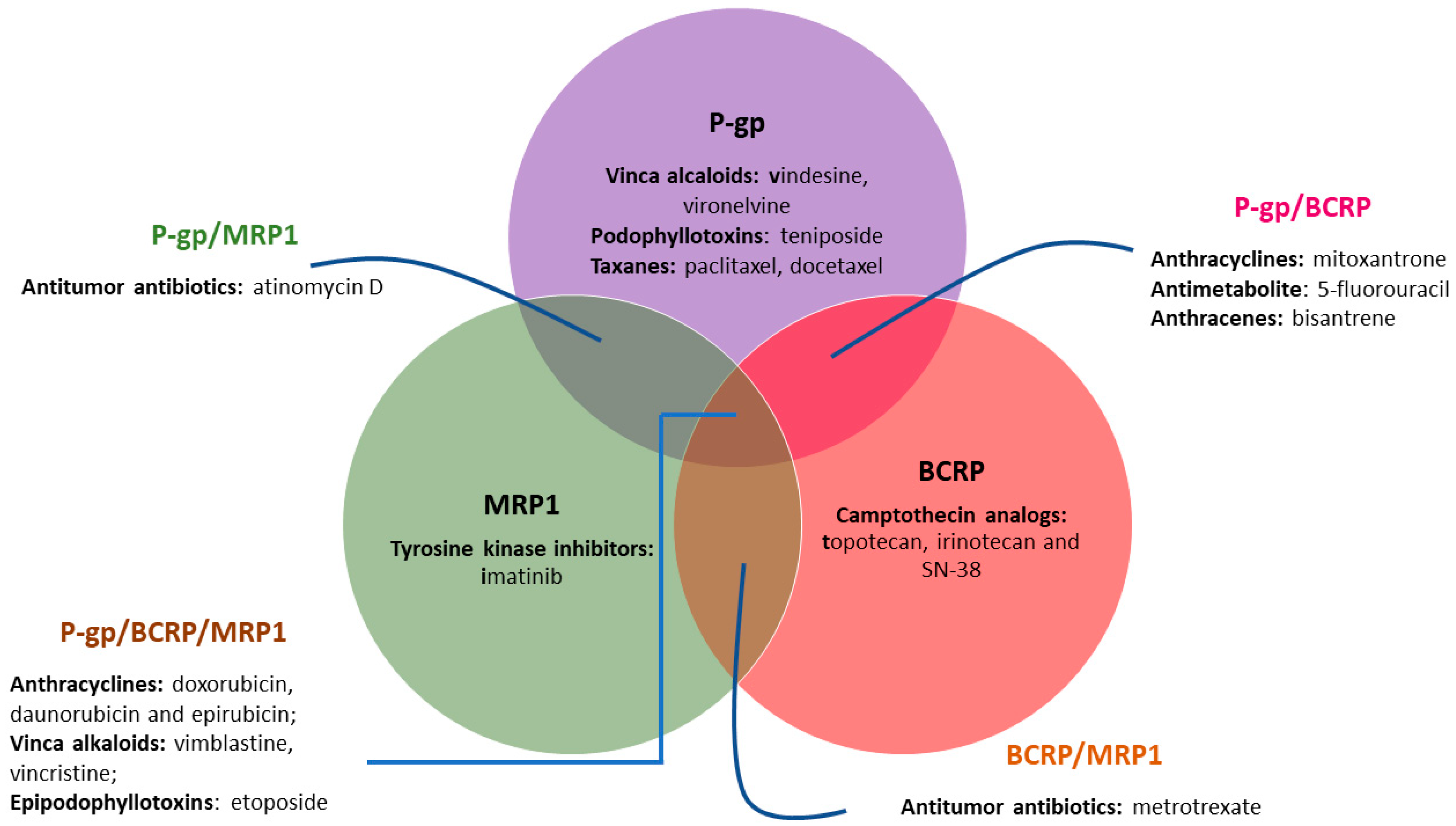
1.1. The Role of ABC Transporter Proteins in Cancer Multidrug Resistance
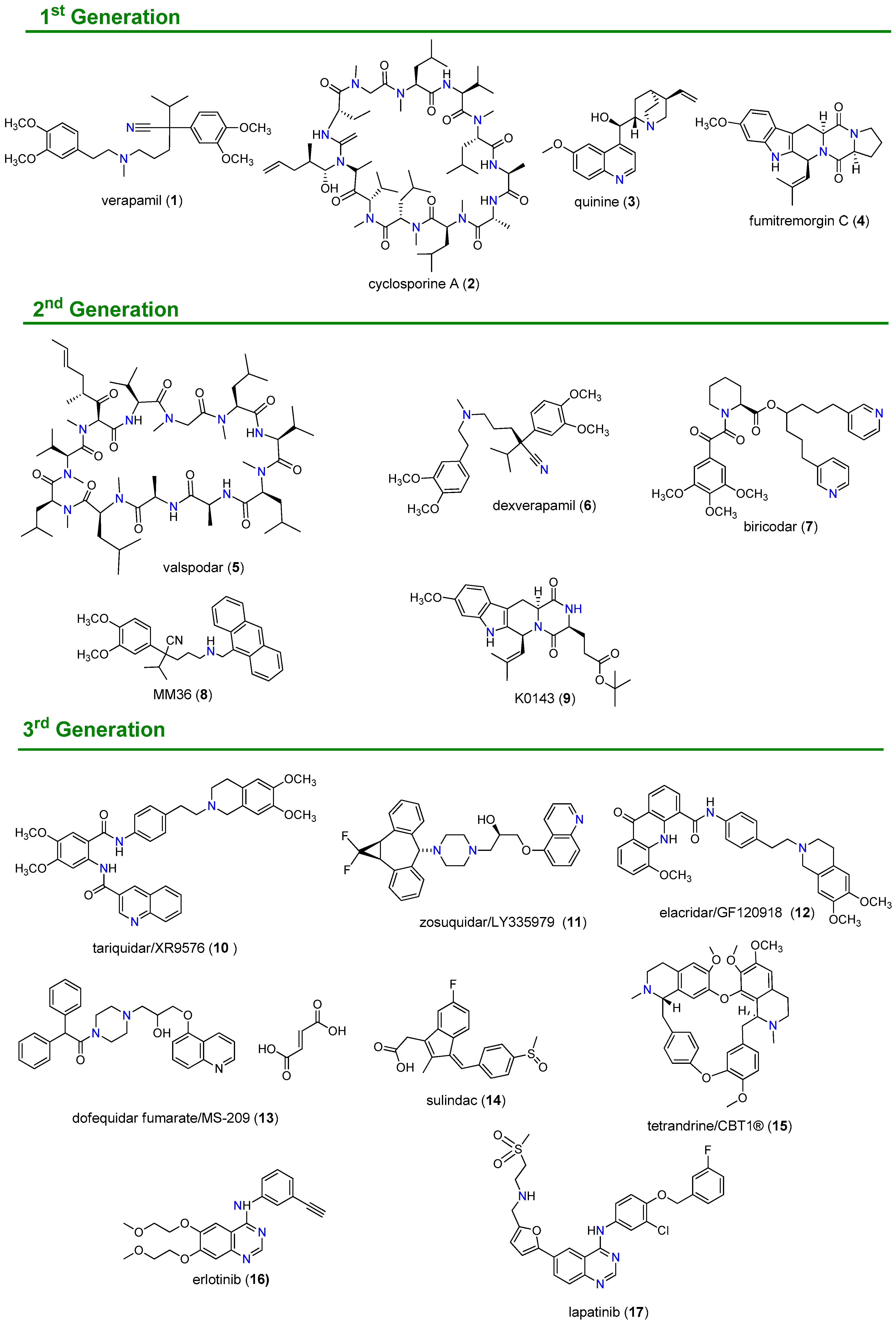
1.2. Inhibition of ABC Transporter Proteins as a Strategy to Overcome MDR
1.3. Natural Products as the Fourth Generation of ABC Transporter Inhibitors
1.4. Nitrogen-Bearing Compounds as Modulators of ABC Transporters
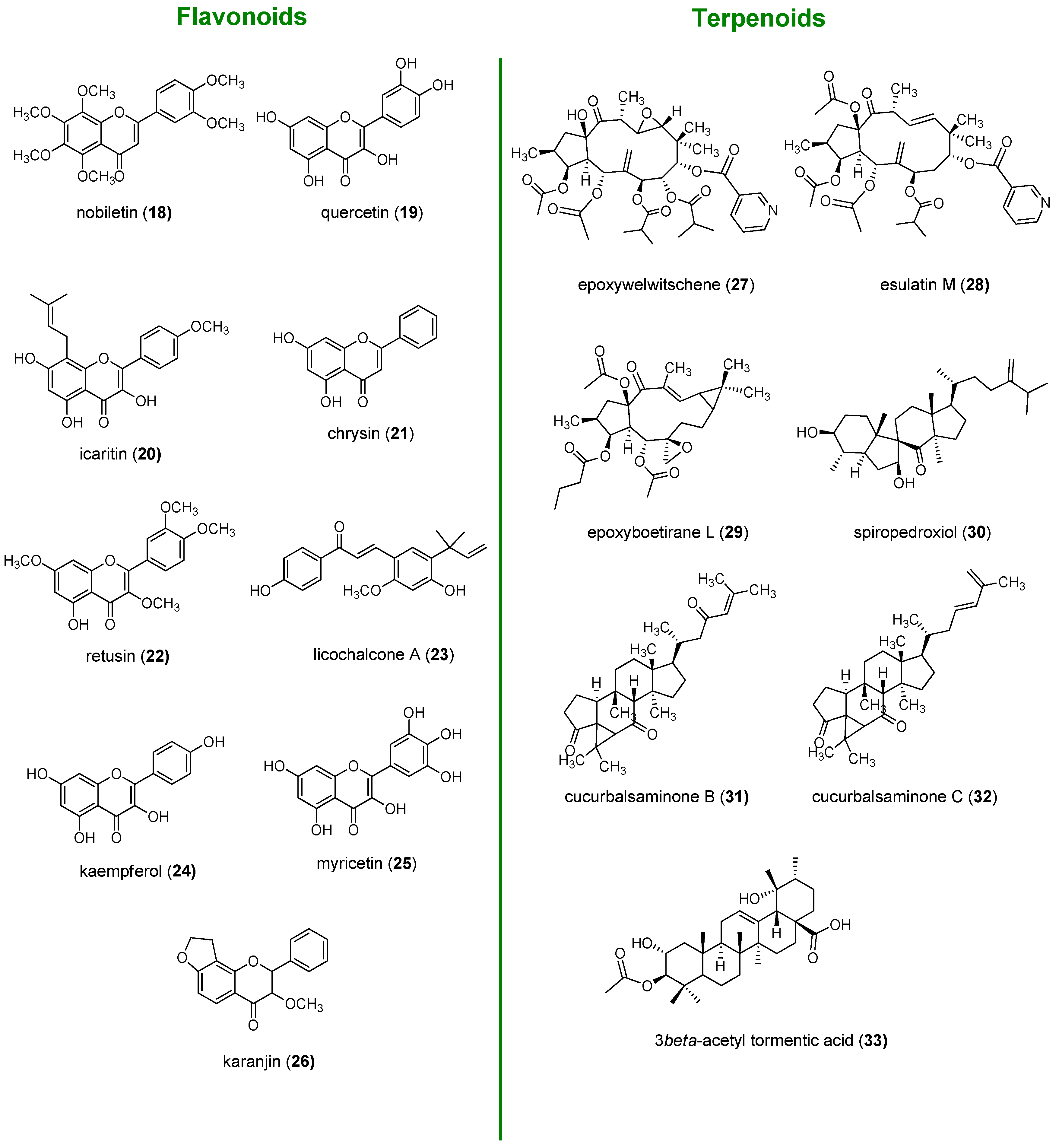
2. Flavonoids
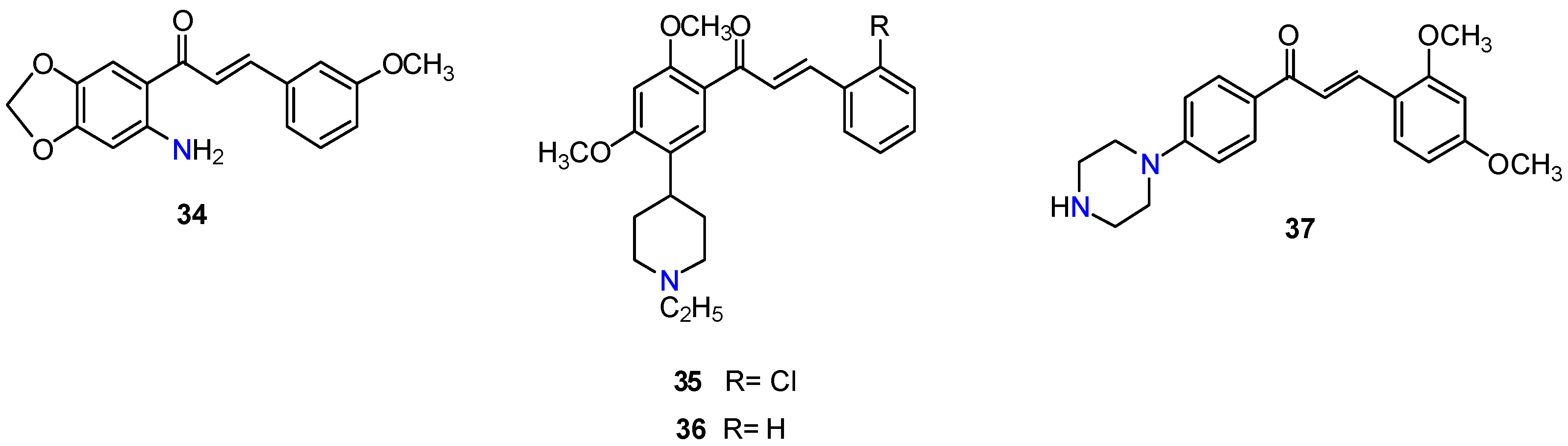
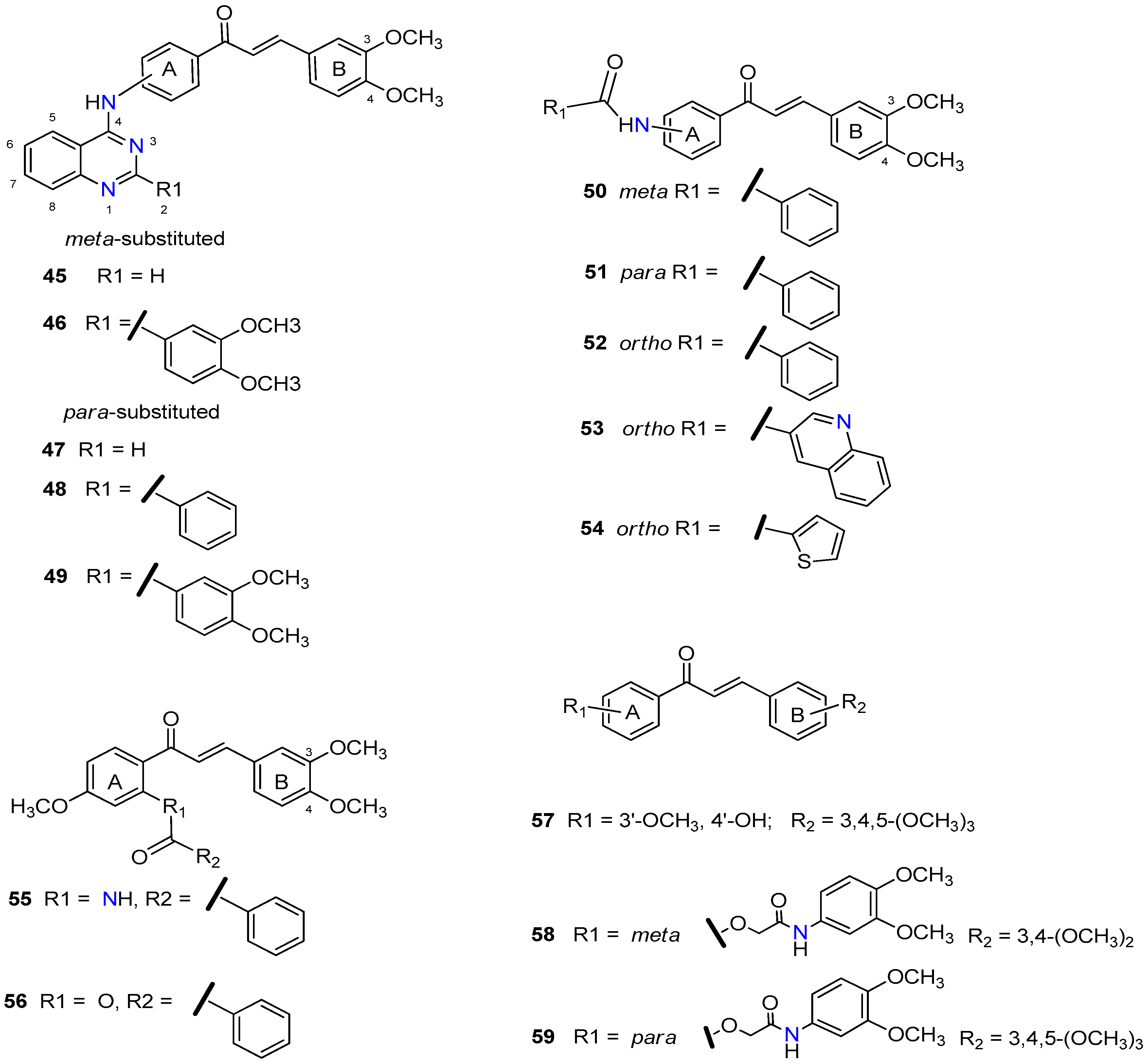
2.1. Chalcones
2.2. Flavanones and Flavanonols
2.3. Flavones
2.4. Flavonols
2.5. Flavanols
3. Terpenes
3.1. Sesquiterpenoids
3.2. Diterpenoids
3.3. Triterpenoids
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.-P.; Gottesman, M.M. Chapter 4 mechanisms of multidrug resistance in cancer. In Multi-Drug Resistance in Cancer. Methods in Molecular Biology (Methods and Protocols); Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 596, pp. 47–75. ISBN 9781607614166. [Google Scholar]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updat. 2019, 46. [Google Scholar] [CrossRef]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The role of eukaryotic and prokaryotic ABC transporter family in failure of chemotherapy. Front. Pharmacol. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2014, 14, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.J.; Ferreira, M.J.U.; dos Santos, D.J.V.A. Reversing cancer multidrug resistance: Insights into the efflux by ABC transports from in silico studies. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 27–55. [Google Scholar] [CrossRef]
- Szöllősi, D.; Rose-Sperling, D.; Hellmich, U.A.; Stockner, T. Comparison of mechanistic transport cycle models of ABC exporters. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 818–832. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; De Lourdes Bastos, M.; Remião, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, Function and Regulation of P-Glycoprotein and Its Clinical Relevance in Drug Disposition. Xenobiotica. 2008, 38, 802–832. [Google Scholar] [CrossRef]
- Seelig, A.; Landwojtowicz, E. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 2000, 12, 31–40. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Schriml, L.M.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.N.D.S.; Ferreira, M.J.U.; Dos Santos, D.J.V.A. Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; To, K.K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 7389, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Stefan, S.M. Multi-target ABC transporter modulators: What next and where to go? Future Med. Chem. 2019, 11, 2353–2358. [Google Scholar] [CrossRef]
- Jaramillo, A.C.; Al Saig, F.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Jamis-Dow, C.; Bryant, G.; Balis, F.M.; Klecker, R.W.; Chabner, S.E.B.B.A.; Steinberg, S.M.; Kohler, D.R.; Wittes, R.E. Phase I and pharmacokinetic study of the multidrug resistance modulator dexverapamil with EPOCH chemotherapy. J. Clin. Oncol. 1995, 13, 1985–1994. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Smith, L.; Wang, Y.M.; Chaturvedi, P.; Villalona, M.; Campbell, E.; Aylesworth, C.; Eckhardt, S.G.; Hammond, L.; Kraynak, M.; et al. Phase I and pharmacokinetic study of paclitaxel in combination with biricodar, a novel agent that reverses multidrug resistance conferred by overexpression of both MDR1 and MRP. J. Clin. Oncol. 1998, 16, 2964–2976. [Google Scholar] [CrossRef] [PubMed]
- Shepard, R.L.; Cao, J.; Starling, J.J.; Dantzig, A.H. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int. J. Cancer 2003, 103, 121–125. [Google Scholar] [CrossRef]
- Tang, R.; Faussat, A.M.; Perrot, J.Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1273–1283. [Google Scholar] [CrossRef]
- Lu, J.F.; Pokharel, D.; Bebawy, M. MRP1 and its role in anticancer drug resistance. Drug Metab. Rev. 2015, 47, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Nomizu, T.; Toi, M.; Ito, Y.; Noguchi, S.; Kobayashi, T.; Asaga, T.; Minami, H.; Yamamoto, N.; Aogi, K.; et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J. Clin. Oncol. 2007, 25, 411–417. [Google Scholar] [CrossRef]
- O’Connor, R.; O’Leary, M.; Ballot, J.; Collins, C.D.; Kinsella, P.; Mager, D.E.; Arnold, R.D.; O’Driscoll, L.; Larkin, A.; Kennedy, S.; et al. A phase I clinical and pharmacokinetic study of the multi-drug resistance protein-1 (MRP-1) inhibitor sulindac, in combination with epirubicin in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 79–87. [Google Scholar] [CrossRef]
- Stefan, S.M.; Wiese, M. Small-molecule inhibitors of multidrug resistance-associated protein 1 and related processes: A historic approach and recent advances. Med. Res. Rev. 2019, 39, 176–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed-Belkacem, A.; Pozza, A.; Macalou, S.; Pérez-Victoria, J.M.; Boumendjel, A.; Di Pietro, A. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2). Anticancer Drugs 2006, 17, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of human ABCG2: From technical Background to recent updates with clinical implications. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, P.; Telu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Halldin, C.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The “specific” P-glycoprotein inhibitor tariquidar is also a substrate and an inhibitor for Breast Cancer Resistance Protein (BCRP/ABCG2). ACS Chem. Neurosci. 2011, 2, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Steadman, K.; Polgar, O.; Morisaki, K.; Blayney, M.; Mistry, P.; Bates, S.E. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004, 64, 1242–1246. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.D.; Brinkhuis, R.F.; Wijnholds, J.; Schinkel, A.H. The mouse Bcrp1/Mxr/Abcp gene: Amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999, 59, 4237–4241. [Google Scholar] [PubMed]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Ghazaly, E.; Kitromilidou, C.; McGrowder, E.H.; Joel, S.; Powles, T. A synergistic interaction between lapatinib and chemotherapy agents in a panel of cell lines is due to the inhibition of the efflux pump BCRP. Mol. Cancer Ther. 2010, 9, 3322–3329. [Google Scholar] [CrossRef] [Green Version]
- Pick, A.; Wiese, M. Tyrosine kinase inhibitors influence ABCG2 expression in EGFR-positive MDCK BCRP cells via the PI3K/Akt signaling pathway. ChemMedChem 2012, 7, 650–662. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, P.; Asenso, J.; Yang, X.D.; Wang, C.; Wei, W. Advances in plant-based inhibitors of P-glycoprotein. J. Enzym. Inhib. Med. Chem. 2016, 31, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E.; Branch, O.; Pike, R. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Harding, M.W.; Neubauer, M.; Langer, C.J.; Moore, M.; Ross, H.J.; Johnson, B.E.; Lynch, T.J. A phase II study of the safety and efficacy of the multidrug resistance inhibitor VX-710 combined with doxorubicin and vincristine in patients with recurrent small cell lung cancer. Cancer 2007, 109, 924–932. [Google Scholar] [CrossRef]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cripe, L.D.; Uno, H.; Paietta, E.M.; Litzow, M.R.; Ketterling, R.P.; Bennett, J.M.; Rowe, J.M.; Lazarus, H.M.; Luger, S.; Tallman, M.S. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: A randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 2010, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Kuppens, I.E.L.M.; Witteveen, E.O.; Jewell, R.C.; Radema, S.A.; Paul, E.M.; Mangum, S.G.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H.M. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin. Cancer Res. 2007, 13, 3276–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John Crown, M. Phase II Trial of the Multi-Drug Resistance Protein Modulating Agent Sulindac in Combination with Epirubicin in Patients with Advanced Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00755976?term=sulindac+and+epirubicin&draw=2&rank=1 (accessed on 8 April 2020).
- Cote, G.M.; Chawla, S.P.; Burgess, M.A.; Thornton, K.A.; Oldham, R.K.; Okuno, S.H.; Ballman, K.V.; Matlow, S.; Barnett, D.; Attia, S. CBT-1 in combination with doxorubicin in patients with metastatic, unresectable sarcomas who previously progressed on doxorubicin. J. Clin. Oncol. 2019, 37, TPS11077. [Google Scholar] [CrossRef]
- Stewart, C.F.; Tagen, M.; Schwartzberg, L.S.; Blakely, L.J.; Tauer, K.W.; Smiley, L.M. Phase I dosage finding and pharmacokinetic study of intravenous topotecan and oral erlotinib in adults with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Weroha, S.J.; Oberg, A.L.; Ziegler, K.L.A.; Dakhilm, S.R.; Rowland, K.M.; Hartmann, L.C.; Moore, D.F.; Keeney, G.L.; Peethambaram, P.P.; Haluska, P. Phase II trial of lapatinib and topotecan (LapTop) in patients with platinum-refractory/resistant ovarian and primary peritoneal carcinoma. Gynecol. Oncol. 2011, 122, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Karthikeyan, S.; Hoti, S. Development of fourth generation abc inhibitors from natural products: A novel approach to overcome cancer multidrug resistance. Anticancer Agents Med. Chem. 2015, 15, 605–615. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.W. Reversal of multidrug resistance in cancer by multi-functional flavonoids. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Paterna, A.; Kincses, A.; Spengler, G.; Mulhovo, S.; Molnár, J.; Ferreira, M.J.U. Dregamine and tabernaemontanine derivatives as ABCB1 modulators on resistant cancer cells. Eur. J. Med. Chem. 2017, 128, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Paterna, A.; Khonkarn, R.; Mulhovo, S.; Moreno, A.; Madeira Girio, P.; Baubichon-Cortay, H.; Falson, P.; Ferreira, M.J.U. Monoterpene indole alkaloid azine derivatives as MDR reversal agents. Bioorg. Med. Chem. 2018, 26, 421–434. [Google Scholar] [CrossRef]
- Nabekura, T.; Yamaki, T.; Kitagawa, S. Effects of chemopreventive citrus phytochemicals on human P-glycoprotein and multidrug resistance protein 1. Eur. J. Pharmacol. 2008, 600, 45–49. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Effect of quercetin in Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem. Pharmacol. 1997, 53, 587–596. [Google Scholar] [CrossRef]
- Sun, L.; Chen, W.; Qu, L.; Wu, J.; Si, J. Icaritin reverses multidrug resistance of HepG2/ADR human hepatoma cells via downregulation of MDR1 and P-glycoprotein expression. Mol. Med. Rep. 2013, 8, 1883–1887. [Google Scholar] [CrossRef] [Green Version]
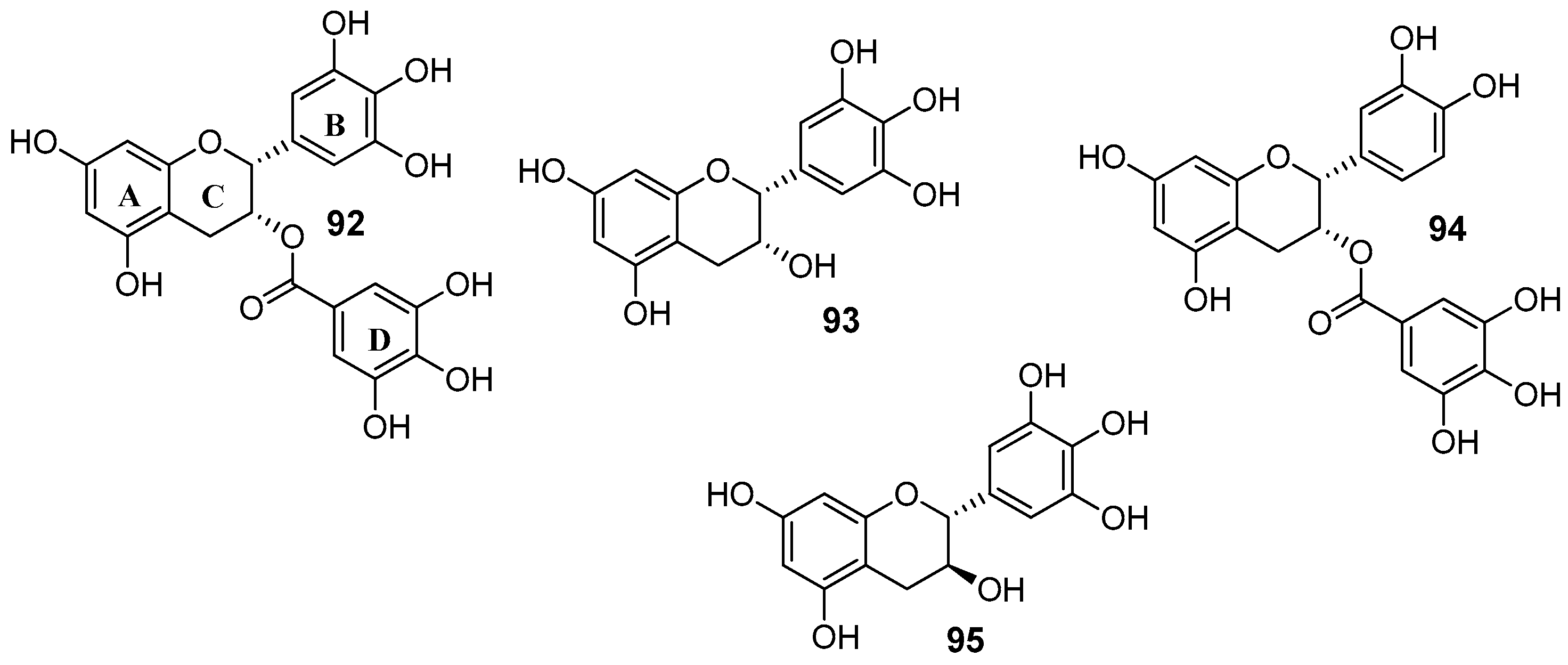
- Pick, A.; Müller, H.; Mayer, R.; Haenisch, B.; Pajeva, I.K.; Weigt, M.; Bönisch, H.; Müller, C.E.; Wiese, M. Structure–activity relationships of flavonoids as inhibitors of breast cancer resistance protein (BCRP). Bioorg. Med. Chem. 2011, 19, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Lusvarghi, S.; Hsiao, S.-H.; Liu, T.-C.; Li, Y.-Q.; Huang, Y.-H.; Hung, T.-H.; Ambudkar, S.V. Licochalcone a selectively resensitizes ABCG2-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. J. Nat. Prod. 2019. [Google Scholar] [CrossRef]
- Juvale, K.; Wiese, M. Design of inhibitors of BCRP/ABCG2. Future Med. Chem. 2015, 7, 1521–1527. [Google Scholar] [CrossRef]
- Leslie, E.M.; Mao, Q.; Oleschuk, C.J.; Deeley, R.G.; Cole, S.P.C. Modulation of multidrug resistance protein 1 (MRP1/ABCC1) transport and ATpase activities by interaction with dietary flavonoids. Mol. Pharmacol. 2001, 59, 1171–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Rothweiler, F.; Nerreter, T.; Sharifi, M.; Ghafourian, T.; Cinatl, J. Karanjin interferes with ABCB1, ABCC1, and ABCG2. J. Pharm. Pharm. Sci. 2014, 17, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalhete, C.; Molnár, J.; Mulhovo, S.; Rosário, V.E.; Ferreira, M.J.U. New potent P-glycoprotein modulators with the cucurbitane scaffold and their synergistic interaction with doxorubicin on resistant cancer cells. Bioorg. Med. Chem. 2009, 17, 6942–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, M.A.; Paterna, A.; Ferreira, R.J.; Lage, H.; Ferreira, M.J.U. Macrocyclic diterpenes resensitizing multidrug resistant phenotypes. Bioorg. Med. Chem. 2014, 22, 3696–3702. [Google Scholar] [CrossRef]
- Ramalhete, C.; Mulhovo, S.; Molnar, J.; Ferreira, M.J.U. Triterpenoids from momordica balsamina: Reversal of ABCB1-mediated multidrug resistance. Bioorg. Med. Chem. 2016, 24, 5061–5067. [Google Scholar] [CrossRef]
- Mónico, A.; Ramalhete, C.; André, V.; Spengler, G.; Mulhovo, S.; Duarte, M.T.; Ferreira, M.J.U. Cucurbalsaminones A−C, rearranged triterpenoids with a 5/6/3/6/5-fused pentacyclic carbon skeleton from momordica balsamina, as multidrug resistance reversers. J. Nat. Prod. 2019, 2138–2143. [Google Scholar] [CrossRef] [Green Version]
- Reis, M.A.; Ahmed, O.B.; Spengler, G.; Molnár, J.; Lage, H.; Ferreira, M.J.U. Jatrophane diterpenes and cancer multidrug resistance—ABCB1 efflux modulation and selective cell death induction. Phytomedicine 2016, 23, 968–978. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.J.U.; Duarte, N.; Reis, M.; Madureira, A.M.; Molnár, J. Euphorbia and Momordica metabolites for overcoming multidrug resistance. Phytochem. Rev. 2014, 13, 915–935. [Google Scholar] [CrossRef]
- Vieira, C.; Duarte, N.; Reis, M.A.; Spengler, G.; Madureira, A.M.; Molnár, J.; Ferreira, M.J.U. Improving the MDR reversal activity of 6,17-epoxylathyrane diterpenes. Bioorg. Med. Chem. 2014, 22, 6392–6400. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.J.; Kincses, A.; Gajdács, M.; Spengler, G.; Dos Santos, D.J.V.A.; Molnár, J.; Ferreira, M.J.U. Terpenoids from euphorbia pedroi as multidrug-resistance reversers. J. Nat. Prod. 2018, 81, 2032–2040. [Google Scholar] [CrossRef]
- Da Graça Rocha, G.; Simões, M.; Oliveira, R.R.; Kaplan, M.A.C.; Gattass, C.R. Effects of 3β-acethyl tormentic acid (3ATA) on ABCC proteins activity. Int. J. Mol. Sci. 2012, 13, 6757–6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Dong, J.; Cai, Y.; Shi, X.; Wang, H.; Liu, G.; Tang, Y.; Liu, J.; Ma, L. Design, synthesis and biological evaluation of chalcones as reversers of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 180, 350–366. [Google Scholar] [CrossRef]
- Krawczyk, S.; Baumert, C.; Hilgeroth, A. Recent developments of P-gp inhibitors. In Advances in Anticancer Agents in Medicinal Chemistry; Prudhomme, M., Ed.; Benthan E books: Sharjah, UAE, 2009; pp. 186–187. ISBN 978-1-60805-712-2. [Google Scholar]
- Zamora, J.M.; Pearce, H.L.; Beck, W.T. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells. Mol. Pharmacol. 1988, 4, 454–462. [Google Scholar]
- Ecker, G.; Huber, M.; Schmid, D.; Chiba, P. The importance of a nitrogen atom in modulators of multidrug resistance. Mol. Pharmacol. 1999, 56, 791–796. [Google Scholar] [PubMed]
- Cramer, J.; Kopp, S.; Bates, S.E.; Chiba, P.; Ecker, G.F. Multispecificity of drug transporters: Probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem 2007, 2, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.; Montanari, F.; Cseke, A.; Wlcek, K.; Visvader, L.; Palme, S.; Chiba, P.; Kuchler, K.; Urban, E.; Ecker, G.F. Subtle structural differences trigger inhibitory activity of propafenone analogues at the two polyspecific ABC transporters: P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). ChemMedChem 2016, 1380–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
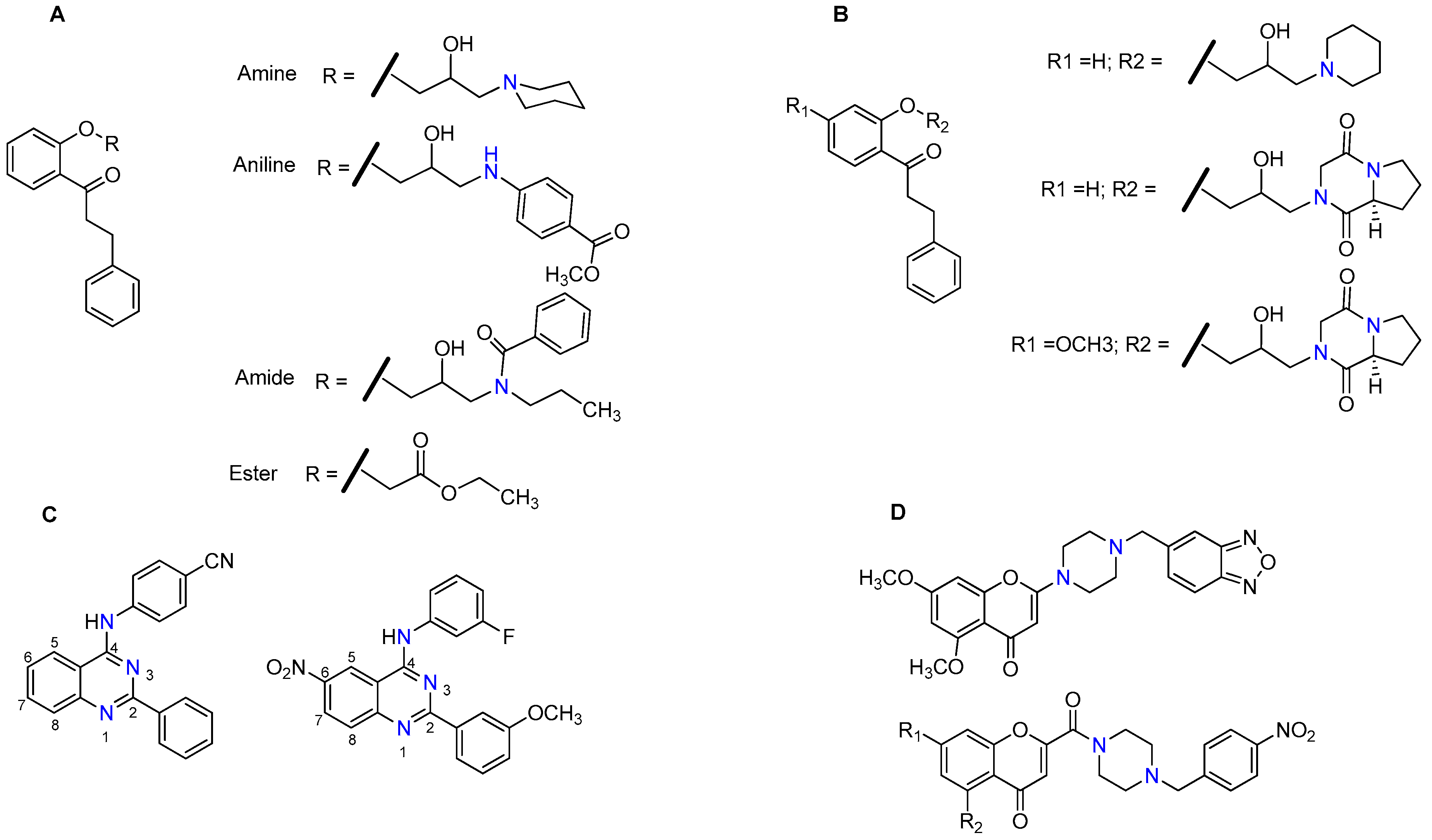
- Krapf, M.K.; Wiese, M. Synthesis and biological evaluation of 4-Anilino-quinazolines and-quinolines as inhibitors of breast cancer resistance protein (ABCG2). J. Med. Chem. 2016, 59, 5449–5461. [Google Scholar] [CrossRef]
- Krapf, M.K.; Gallus, J.; Namasivayam, V.; Wiese, M. 2,4,6-Substituted quinazolines with extraordinary inhibitory potency toward ABCG2. J. Med. Chem. 2018, 61, 7952–7976. [Google Scholar] [CrossRef]
- Obreque-Balboa, J.E.; Sun, Q.; Bernhardt, G.; König, B.; Buschauer, A. Flavonoid derivatives as selective ABCC1 modulators: Synthesis and functional characterization. Eur. J. Med. Chem. 2016, 109, 124–133. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Kumar, A.; Malik, A.K. Flavonoids biosynthesis in plants and its further analysis by capillary electrophoresis. Electrophoresis 2017, 38, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Dewick, P.M. Medicinal Natural Products—A Biosynthetic Approach, 3rd ed.; Dewick, P.M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; ISBN 978-0-470-74168-9. [Google Scholar]
- Gupta, V.K.; Bhalla, Y.; Jaitak, V. Impact of ABC transporters, glutathione conjugates in MDR and their modulation by flavonoids: An overview. Med. Chem. Res. 2014, 23, 1–15. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.Y.; Xia, P.; Bastow, K.F.; Nakanishi, Y.; Lee, K.H. Antitumor agents. Part 202: Novel 2′-amino chalcones: Design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2000, 10, 699–701. [Google Scholar] [CrossRef]
- Liu, X.L.; Tee, H.W.; Go, M.L. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg. Med. Chem. 2008, 16, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Winter, E.; Gozzi, G.J.; Chiaradia-Delatorre, L.D.; Daflon-Yunes, N.; Terreux, R.; Gauthier, C.; Mascarello, A.; Lleal, P.C.; Cadena, S.M.; Yunes, R.A.; et al. Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2: Polyspecificity at B-ring position. Drug Des. Dev. Ther. 2014, 8, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Kraege, S.; Köhler, S.C.; Wiese, M. Acryloylphenylcarboxamides: A new class of breast cancer resistance protein (ABCG2) modulators. ChemMedChem 2016, 11, 2422–2435. [Google Scholar] [CrossRef]
- Kraege, S.; Stefan, K.; Köhler, S.C.; Wiese, M. Optimization of acryloylphenylcarboxamides as inhibitors of ABCG2 and comparison with acryloylphenylcarboxylates. ChemMedChem 2016, 11, 2547–2558. [Google Scholar] [CrossRef]
- Silbermann, K.; Shah, C.P.; Sahu, N.U.; Juvale, K.; Stefan, S.M.; Kharkar, P.S.; Wiese, M. Novel chalcone and flavone derivatives as selective and dual inhibitors of the transport proteins ABCB1 and ABCG2. Eur. J. Med. Chem. 2019, 164, 193–213. [Google Scholar] [CrossRef]
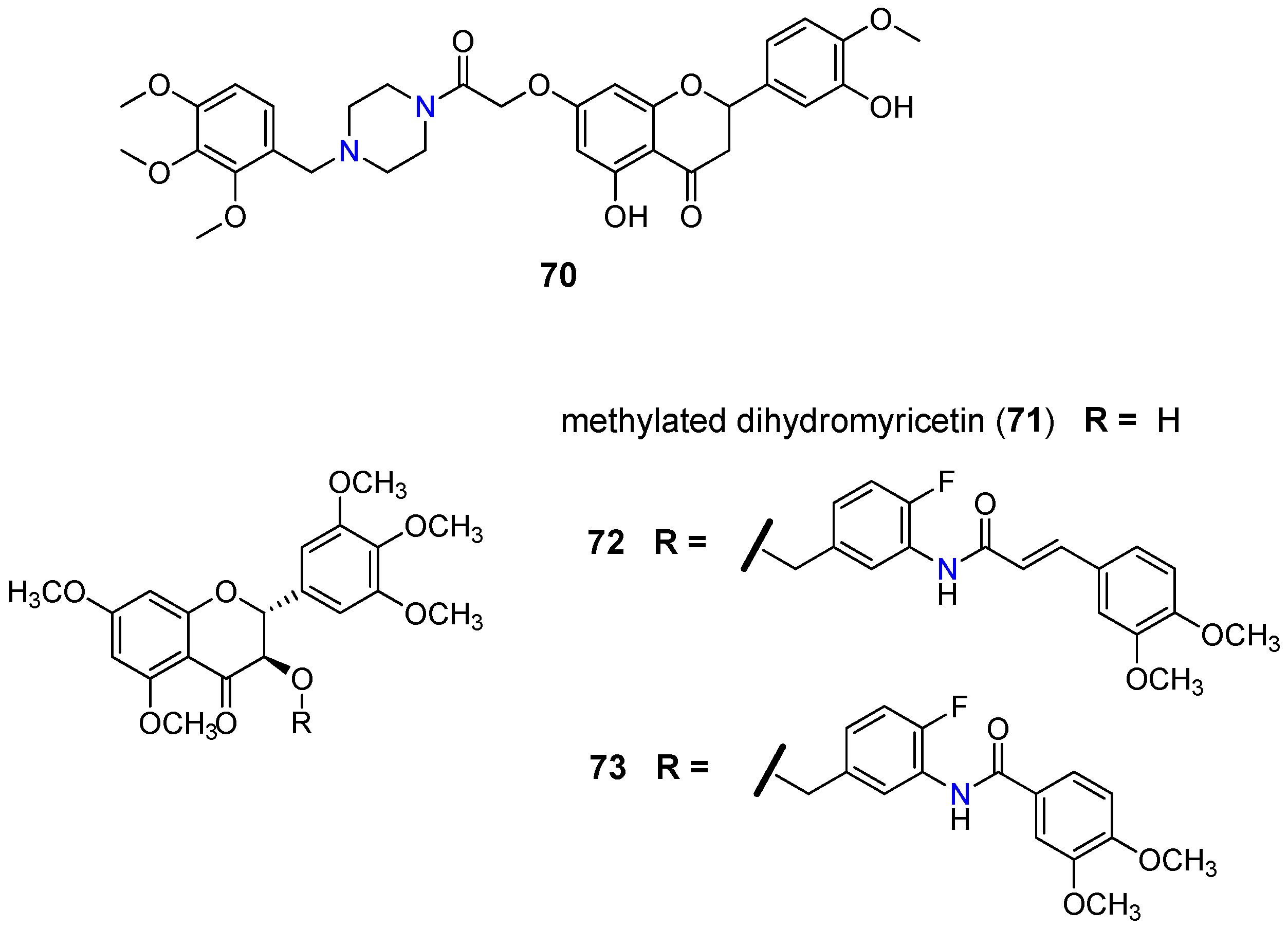
- Ferreira, R.J.; Baptista, R.; Moreno, A.; Madeira, P.G.; Khonkarn, R.; Baubichon-Cortay, H.; Dos Santos, D.J.; Falson, P.; Ferreira, M.J.U. Optimizing the flavanone core toward new selective nitrogen-containing modulators of ABC transporters. Future Med. Chem. 2018, 10, 725–741. [Google Scholar] [CrossRef]
- Ferté, J.; Kühnel, J.M.; Chapuis, G.; Rolland, Y.; Lewin, G.; Schwaller, M.A. Flavonoid-related modulators of multidrug resistance: Synthesis, pharmacological activity, and structure-activity relationships. J. Med. Chem. 1999, 42, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Kühnel, J.M.; Perrot, J.Y.; Faussat, A.M.; Marie, J.P.; Schwaller, M.A. Functional assay of multidrug resistant cells using JC-1, a carbocyanine fluorescent probe. Leukemia 1997, 11, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, I.L.K.; Wang, B.C.; Yuan, J.; Duan, L.X.; Liu, Z.; Liu, T.; Li, X.M.; Hu, X.; Zhang, X.Y.; Jiang, T.; et al. Potent and nontoxic chemosensitizer of P-glycoprotein-mediated multidrug resistance in cancer: Synthesis and evaluation of methylated epigallocatechin, gallocatechin, and dihydromyricetin derivatives. J. Med. Chem. 2015, 58, 4529–4549. [Google Scholar] [CrossRef] [PubMed]
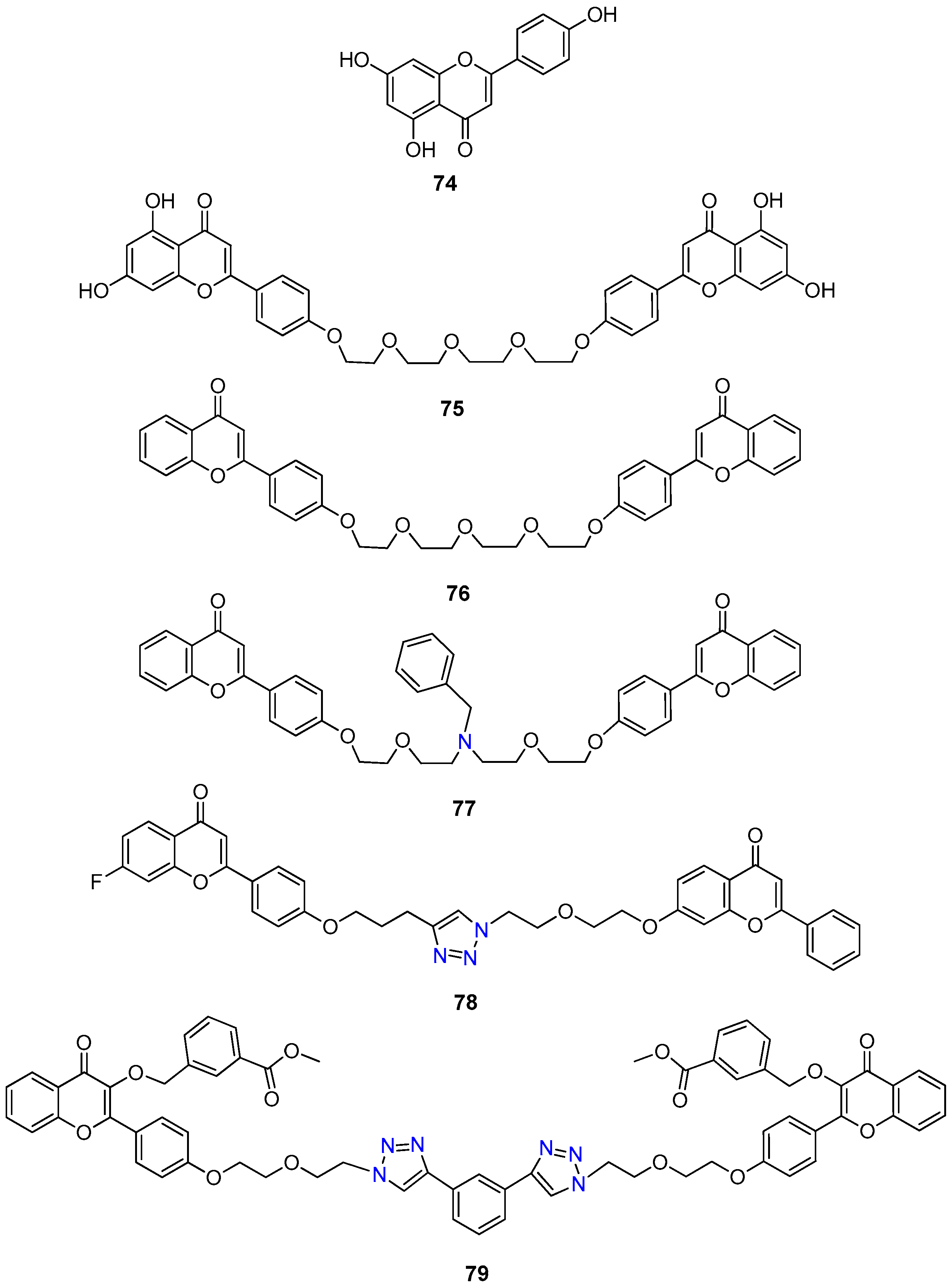
- Yan, C.S.W.; Wong, I.L.K.; Chan, K.-F.; Kan, J.W.Y.; Chong, T.C.; Law, M.C.; Zhao, Y.; Chan, S.W.; Chan, T.H.; Chow, L.M.C. A new class of safe, potent, and specific P-gp modulator: Flavonoid dimer FD18 reverses P-gp-mediated multidrug resistance in human breast xenograft in vivo. Mol. Pharm. 2015, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Zhao, Y.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J. Med. Chem. 2006, 49, 6742–6759. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Zhao, Y.; Chow, T.W.S.; Yan, C.S.W.; Ma, D.L.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Structure-activity relationships. ChemMedChem 2009, 4, 594–614. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Wong, I.L.K.; Kan, J.W.Y.; Yan, C.S.W.; Chow, L.M.C.; Chan, T.H. Amine linked flavonoid dimers as modulators for P-glycoprotein-based multidrug resistance: Structure-activity relationship and mechanism of modulation. J. Med. Chem. 2012, 55, 1999–2014. [Google Scholar] [CrossRef]
- Wong, I.L.K.; Zhu, X.; Chan, K.F.; Law, M.C.; Lo, A.M.Y.; Hu, X.; Chow, L.M.C.; Chan, T.H. Discovery of novel flavonoid dimers to reverse multidrug resistance protein 1 (MRP1, ABCC1) mediated drug resistance in cancers using a high throughput platform with “click Chemistry”. J. Med. Chem. 2018, 61, 9931–9951. [Google Scholar] [CrossRef]
- Zhu, X.; Wong, I.L.K.; Chan, K.-F.; Cui, J.; Law, M.C.; Chong, T.C.; Hu, X.; Chow, L.M.C.; Chan, T.H. Triazole bridged flavonoid dimers as potent, nontoxic, and highly selective breast cancer resistance protein (BCRP/ABCG2) inhibitors. J. Med. Chem. 2019, 62, 8578–8608. [Google Scholar] [CrossRef]
- Dei, S.; Romanelli, M.N.; Manetti, D.; Chiaramonte, N.; Coronnello, M.; Salerno, M.; Teodori, E. Design and synthesis of aminoester heterodimers containing flavone or chromone moieties as modulators of P-glycoprotein-based multidrug resistance (MDR). Bioorg. Med. Chem. 2018, 26, 50–64. [Google Scholar] [CrossRef]
- Ay, M.; Charli, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Quercetin. Nutraceuticals Effic. Saf. Toxic. 2016, 447–452. [Google Scholar] [CrossRef]
- Shebeko, S.K.; Zupanets, I.A.; Popov, O.S.; Tarasenko, O.O.; Shalamay, A.S. Chapter 27—Effects of Quercetin and Its Combinations on Health. In Polyphenols: Mechanisms of Action in Human Health and Disease, 2nd ed.; Elsevier Inc.: San Diego, CA, USA, 2018; pp. 373–394. ISBN 9780128130063. [Google Scholar]
- Miltonprabu, S. Chapter 2.9—Quercetin: A Flavonol with Versatile Therapeutic Applications and Its Interactions with Other Drugs. In Nonvitamin and Nonmineral Nutritional Supplements; Elsevier Inc.: San Diego, CA, USA, 2019; pp. 75–89. ISBN 9780128124918. [Google Scholar]
- Kim, M.K.; Choo, H.; Chong, Y. Water-soluble and cleavable quercetin-amino acid conjugates as safe modulators for P-glycoprotein-based multidrug resistance. J. Med. Chem. 2014, 57, 7216–7233. [Google Scholar] [CrossRef]
- Balentine, D.A.; Wiseman, S.A.; Bouwens, L.C.M. The chemistry of tea flavonoids. Crit. Rev. Food Sci. Nutr. 1997, 37, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Sawai, Y.; Yamaguchi, Y. Simultaneous analysis of catechins, gallic acid, strictinin, and purine alkaloids in green tea by using catechol as an internal standard. J. Agric. Food Chem. 2007, 55, 4957–4964. [Google Scholar] [CrossRef] [PubMed]
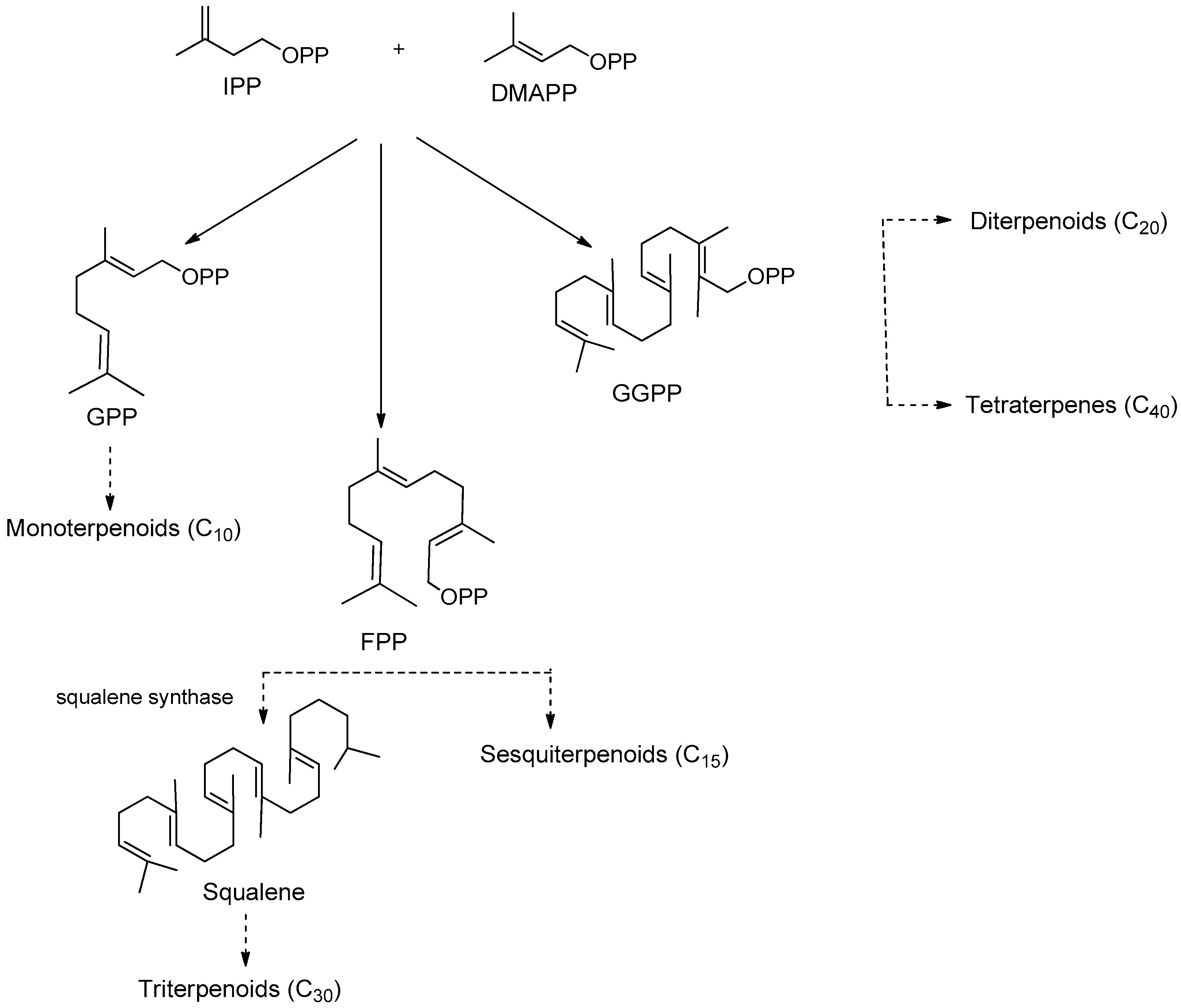
- Liby, K.T.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 7, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Sawai, S.; Saito, K. Triterpenoid biosynthesis and engineering in plants. Front. Plant Sci. 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.X.; Lou, Y.G.; Mao, Y.B.; Lu, S.; Wang, L.J.; Chen, X.Y. Plant terpenoids: Biosynthesis and ecological functions. J. Integr. Plant Biol. 2007, 49, 179–186. [Google Scholar] [CrossRef]
- Yan, X.J.; Gong, L.H.; Zheng, F.Y.; Cheng, K.J.; Chen, Z.S.; Shi, Z. Triterpenoids as reversal agents for anticancer drug resistance treatment. Drug Discov. Today 2014, 19, 482–488. [Google Scholar] [CrossRef]
- Matos, A.M.; Reis, M.; Duarte, N.; Spengler, G.; Molnár, J.; Ferreira, M.J.U. Epoxylathyrol derivatives: Modulation of ABCB1-mediated multidrug resistance in human colon adenocarcinoma and mouse T-lymphoma cells. J. Nat. Prod. 2015, 78, 2215–2228. [Google Scholar] [CrossRef]
- Reis, M.A.; Ahmed, O.B.; Spengler, G.; Molnár, J.; Lage, H.; Ferreira, M.J.U. Exploring jolkinol D derivatives to overcome multidrug resistance in cancer. J. Nat. Prod. 2017, 80, 1411–1420. [Google Scholar] [CrossRef]
- Reis, M.A.; Matos, A.M.; Duarte, N.; Ahmed, O.B.; Ferreira, R.J.; Lage, H.; Ferreira, M.J.U. Epoxylathyrane derivatives as MDR-selective compounds for disabling multidrug resistance in cancer. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.P.; Muñoz-Martínez, F.; Torrecillas, I.R.; Mendoza, C.R.; Gamarro, F.; Bazzocchi, I.L.; Núñez, M.J.; Pardo, L.; Castanys, S.; Campillo, M.; et al. Biological evaluation, structure-activity relationships, and three-dimensional quantitative structure-activity relationship studies of dihydro-β-agarofuran sesquiterpenes as modulators of P-glycoprotein-dependent multidrug resistance. J. Med. Chem. 2007, 50, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Callies, O.; Sánchez-Cañete, M.P.; Gamarro, F.; Jiménez, I.A.; Castanys, S.; Bazzocchi, I.L. Optimization by molecular fine tuning of dihydro-β-agarofuran sesquiterpenoids as reversers of P-glycoprotein-mediated multidrug resistance. J. Med. Chem. 2016, 59, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Zhang, D.M.; Shu, C.; Chen, J.J.; Sodani, K.; Wang, J.; Bhatnagar, J.; Lan, P.; Ruan, Z.X.; Xiao, Z.J.; Ambudkar, S.V.; et al. BBA, a derivative of 23-hydroxybetulinic acid, potently reverses ABCB1-mediated drug resistance in vitro and in vivo. Mol. Pharm. 2012, 9, 3147–3159. [Google Scholar] [CrossRef]
- Zhang, D.M.; Li, Y.J.; Shu, C.; Ruan, Z.X.; Chen, W.M.; Yiu, A.; Peng, Y.H.; Wang, J.; Lan, P.; Yao, Z.; et al. Bipiperidinyl derivatives of 23-hydroxybetulinic acid reverse resistance of HepG2/ADM and MCF-7/ADR cells. Anticancer Drugs 2013, 24, 441–454. [Google Scholar] [CrossRef]
- Braga, F.; Ayres-Saraiva, D.; Gattass, C.R.; Capella, M.A.M. Oleanolic acid inhibits the activity of the multidrug resistance protein ABCC1 (MRP1) but not of the ABCB1 (P-glycoprotein): Possible use in cancer chemotherapy. Cancer Lett. 2007, 248, 147–152. [Google Scholar] [CrossRef]
- Villar, V.H.; Vögler, O.; Barceló, F.; Gómez-Florit, M.; Martínez-Serra, J.; Obrador-Hevia, A.; Martín-Broto, J.; Ruiz-Gutiérrez, V.; Alemany, R. Oleanolic and maslinic acid sensitize soft tissue sarcoma cells to doxorubicin by inhibiting the multidrug resistance protein MRP-1, but not P-glycoprotein. J. Nutr. Biochem. 2014, 25, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Paszel-Jaworska, A.; Rubiś, B.; Bednarczyk-Cwynar, B.; Zaprutko, L.; Rybczyńska, M. Proapoptotic activity and ABCC1-related multidrug resistance reduction ability of semisynthetic oleanolic acid derivatives DIOXOL and HIMOXOL in human acute promyelocytic leukemia cells. Chem. Biol. Interact. 2015, 242. [Google Scholar] [CrossRef]
- Ai, Y.; Kang, F.; Huang, Z.; Xue, X.; Lai, Y.; Peng, S.; Tian, J.; Zhang, Y. Synthesis of CDDO-amino acid-nitric oxide donor trihybrids as potential antitumor agents against both drug-sensitive and drug-resistant colon cancer. J. Med. Chem. 2015, 58, 2452–2464. [Google Scholar] [CrossRef]
- Zhu, B.; Ren, C.; Du, K.; Zhu, H.; Ai, Y.; Kang, F.; Luo, Y.; Liu, W.; Wang, L.; Yang, X.; et al. Olean-28,13b-olide 2 plays a role in cisplatin-mediated apoptosis and reverses cisplatin resistance in human lung cancer through multiple signaling pathways. Biochem. Pharmacol. 2019, 170. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guan, D.; Lei, L.; Lu, J.; Liu, J.Q.; Yang, G.; Yan, C.; Zhai, R.; Tian, J.; Bi, Y.; et al. H6, a novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol. Appl. Pharmacol. 2018, 341, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, Q.; Liu, X.; Yang, Y.; Wang, B.; Zhai, R.; Qi, J.G.; Tian, J.; Wang, H.; Bi, Y. Synthesis and biological evaluation of novel H6 analogues as drug resistance reversal agents. Eur. J. Med. Chem. 2019, 161, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Huang, T.; Xue, M.; Chen, J.; Feng, L.; Du, R.; Feng, Y. Current knowledge and development of hederagenin as a promising medicinal agent: A comprehensive review. RSC Adv. 2018, 8, 24188–24202. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Baek, S.; Shin, D.; Lee, J.; Roh, J.L. Hederagenin induces apoptosis in cisplatin-resistant head and neck cancer cells by inhibiting the Nrf2-ARE antioxidant pathway. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Wang, X.; Wang, K.; Liu, J.; Lei, L.; Luo, X.; Zhai, R.; Fu, F.; Wang, H.; et al. Design, synthesis and biological evaluation of novel α-hederagenin derivatives with anticancer activity. Eur. J. Med. Chem. 2017, 141, 427–439. [Google Scholar] [CrossRef]
- Zhao, Y.; Bu, L.; Yan, H.; Jia, W. 20S-protopanaxadiol inhibits P-glycoprotein in multidrug resistant cancer cells. Planta Med. 2009, 75, 1124–1128. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Liu, P.; Deng, R.; Lei, M.; Chen, W.; Hu, L. 20(S)-Protopanaxadiol (PPD) analogues chemosensitize multidrug-resistant cancer cells to clinical anticancer drugs. Bioorg. Med. Chem. 2013, 21, 4279–4287. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liu, J.; Chen, W.; Xu, Q.; Xiao, M.; Hu, L.; Mao, L.; Wang, X. A 20(S)-protopanoxadiol derivative overcomes multi-drug resistance by antagonizing ATP-binding cassette subfamily B member 1 transporter function. Oncotarget 2016, 7, 9388–9403. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Chen, W.; Hu, L.; Pan, J.; Wang, X. A 20(S)-protopanaxadiol derivative PPD12 reverse ABCB1-mediated multidrug resistance with oral bioavailability and low toxicity. Oncol. Lett. 2018, 16, 5891–5899. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.K.; Zhang, H.; Zhang, G.N.; Wang, Y.J.; Kathawala, R.J.; Si, R.; Patel, B.A.; Xu, J.; Chen, Z.S. Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents. Oncotarget 2015, 6, 24277–24290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Yang, G.; Guo, M.; Guo, J.; Li, Y.; Lu, J.; Yang, Q.; Tang, H.; Li, Y.; Fang, X.; et al. Design, synthesis, and discovery of ocotillol-type amide derivatives as orally available modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 161, 118–130. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target Protein | Clinical Trial Phase | State | Ref. |
|---|---|---|---|---|
| Valspodar (PSC 833, 5) | P-gp | III | Completed 1997–2000 | [53] |
| Biricodar (VX-710, 7) | P-gp/MRP1 | II | Terminated 1998–2001 | [54] |
| Tariquidar (XR9576, 10) | P-gp | II | Completed 2003–2009 | [55] |
| Zozuquidar (LY335979, 11) | P-gp | III | Completed 2002–2009 | [56] |
| Elacridar (GF120918, 12) | BCRP | I | Completed 2002–2004 | [57] |
| Dofequidar (MS-209, 13) | P-gp/MRP1 | III | Completed | [40] |
| Sulindac (14) | MRP1 | II | Completed 2008–2012 | [58] |
| Tetrandrine/CBT1® (15) | P-gp | I | Ongoing. Start in March 2018 | [59] |
| Erlotinib (16) | BCRP | I | Completed 2006–2009 | [60] |
| Lapatinib (17) | BCRP | II | Completed 2007–2009 | [61] |
| Compound (Quinoxaline B Ring) | IC50 (µM) 1 | Compound (2-naphthyl B Ring) | IC50 (µM) 1 | Compound (3,4-methylene-dioxyphenyl B Ring) | IC50 (µM) 1 |
|---|---|---|---|---|---|
 | 1.7 |  | 17.0 |  | 5.6 |
 | 1.9 |  | 4.1 |  | 3.5 |
 | 1.4 | - | - |
| Compound | Cell Line and Assay/IC50 1 (µM) | Cell Line/GI50 2 | Ref. | |||
|---|---|---|---|---|---|---|
| MDCK II BCRP Pheo. A | MDCK II BCRP Hoechst 33342 | P-gp Overexpressing A2780adr Calcein AM | MDCK II Wild Type | BDCK II BCRP | ||
| 45 | 1.30 | - | 0.42 | 3.42 | 5.14 | [100] |
| 46 | 0.60 | - | 0.48 | 12.60 | 10.90 | [100] |
| 47 | 0.84 | - | 2.34 | - | - | [100] |
| 48 | 0.29 | - | 18.8 | 3.46 | 4.80 | [100] |
| 49 | 0.19 | - | 14.9 | 132 | 92.9 | [100] |
| 50 | 2.18 | - | - | - | - | [101] |
| 51 | 1.30 | - | - | 7.88 | 7.08 | [101] |
| 52 | 0.98 | 0.50 | - | 17.40 | 20.50 | [101] |
| 53 | 0.97 | 0.57 | 0.49 | 69.30 | 65.90 | [101] |
| 54 | 0.60 | 0.50 | - | 89.40 | 93.20 | [101] |
| 55 | - | 0.22 | 1.13 | 80.00 | 78.00 | [102] |
| 56 | - | 0.88 | - | - | - | [102] |
| 57 | 1.97 | - | - | 7.56 | 9.38 | [103] |
| 58 | 6.33 | - | - | - | - | [103] |
| 59 | 3.37 | - | - | 18.90 | 27.7 | [103] |
| Ko143 (9) | 0.2 | 0.22 | - | 11.10 | 10.9 | [100,101] |
| cyclosporine A (2) | - | - | 1.41 | [100] | ||

| Compound | R1 | R2 | GI50 DOX (µM) 1 | RF 2 |
|---|---|---|---|---|
| 60 |  |  | 23.59 | 2.17 |
| 61 | |  | 1.02 | 50.19 |
| 62 | |  | 14.84 | 3.45 |
| 63 | |  | 31.60 | 1.62 |
| 64 | OH |  | 2.63 | 19.46 |

| Compound | R | Inhibition (%) | |
|---|---|---|---|
| BCRP | MRP1 | ||
| Naringenin (65) | - | 26.6 | 8.91 |
| 66 |  | −2.77 | 62.6 |
| 67 |  | −0.05 | 95.0 |
| 68 |  | 53.5 | 56.3 |
| 69 |  | 71.1 | 45.1 |
| Ko143 (9) (1 µM) | - | 100 | - |
| Verapamil (1) (35 µM) | - | - | 100 |
| Compound | Cell Line/ABC Protein-Mediated Resistance EC50 (µM) 1 | Cell Line /GI50 (µM) 2 | Ref. | |||
|---|---|---|---|---|---|---|
| LCC6MDR P-gp/Paclitaxel Resistance | MCF7-MX100 BCRP/Topotecan Resistance | HEK293/R2 BCRP/Topotecan Resistance | 2008/MRP1 Doxorubicin Resistance | L929 | ||
| 75 | 0.950 | - | - | - | >100 | [108,109] |
| 76 | 0.222 | - | - | - | >100 | [108,110] |
| 77 | 0.148 | - | - | - | 85.000 | [108,111] |
| 78 | - | - | - | 0.053 | >100 | [112] |
| 79 | >1000 | 0.001 | 0.002 | >1000 | >100 | [113] |
| Verapamil (1) | 0.428 | - | - | - | 89.200 | [111] |
| Kol143 (9) | 1.060 | 0.009 | 0.009 | 1.95 | 31.400 | [113] |

| Compound | R1 | R2 | R3 | [I]0.5 (µM) 1 | αmax 2 |
|---|---|---|---|---|---|
| 80 | (CH2)5 | CH3 | OH | 0.96 | 0.99 |
| 81 | (CH2)5 | CH3 | H | 0.34 | 0.99 |
| 82 | (CH2)5 |  | H | 0.43 | 0.99 |
| 83 | (CH2)5 | H | H | 1.32 | 0.69 |
| 84 | (CH2)2O(CH2)2 | CH3 | H | 1.23 | 0.86 |
| Verapamil (1) | - | - | - | 1.60 | 0.70 |

| Compound | IC50 (µM) 1 | EC50 (µM) 2 | GI50 (µM) 3 | Therapeutic Ratio 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MDCK II BCRP | A2780 adr (P-gp) | MDCK II BCRP | A2780 adr (P-gp) | MDCK II wt | BDCK II BCRP | A2780 | A2780/ADR | BCRP | P-gp | |
| 85 | - | 1.89 | - | 5.01 | - | - | 68.7 | 98.6 | - | 52.2 |
| 86 | 5.09 | 1.41 | 0.793 | - | >100 | >100 | - | - | >19.6 | - |
| 87 | 3.75 | 5.43 | 0.446 | 3.12 | >100 | >100 | 43.6 | 45.3 | >26.7 | 8.3 |

| Compound | R1 | R2 | IC50 (µM) 1 |
|---|---|---|---|
| 88 |  | H | 0.41 |
| 89 |  | H | 0.14 |
| 90 |  |  | 0.78 |
| 91 |  |  | 0.71 |

| Compound | R | RF 1 | ||
|---|---|---|---|---|
| LCC6MDR | HEK293/R2 | 2008/MRP1 | ||
| epigallocatechin-3-gallate (92) | - | 1.2 | - | - |
| Epigallocatechin Derivatives | ||||
| 96 |  | 48.2 | 2.7 | 0.8 |
| 97 |  | 43.8 | 2.3 | 0.9 |
| Gallocatechin Derivatives | ||||
| 98 |  | 31.4 | 10.4 | 2.6 |
| 99 |  | 53.6 | 12.3 | 2.6 |
| Verapamil (1) | 3.8 | - | - | |
| K0143 (9) | - | 19.5 | - | |

| Compound | R1 | R2 | Activity | ||
|---|---|---|---|---|---|
| Ki 1 (µM) | RF 2 (Daunomycin) | RF 2 (Vimblastine) | |||
| 100 | OH | OH | 0.28 | 5.6 | 23.7 |
| 101 | OH |  | 0.54 | 10.8 | 19.1 |
| 102 | OH |  | 0.13 | 11.8 | 17.4 |
| 103 | OH |  | 1.21 | 11.8 | 11.2 |
| 104 | OH |  | 0.19 | 15.8 | 27.4 |
| 105 |  | OH | 0.63 | 10.0 | 20.6 |
| Verapamil (1) | - | - | 2.8 | 8.7 | |

| Compound at 20 µM | R | Fluorescence Activity Ratio (FAR) 1 |
|---|---|---|
| Helioscopinolide E (106) | - | 10.23 |
| 107 | H | 4.88 |
| 108 |  | 38.64 |
| 109 |  | 56.37 |
| 110 |  | 40.69 |
| verapamil (1) | - | 9.66 |

| Compound | R1 | R2 | R3 | Cells/In Vivo Models | Biological Activity |
|---|---|---|---|---|---|
| 112 | OH |  | OH | HepG2/ADM | At 5 µM reduced the GI50 1 of vincristine 132-fold and paclitaxel 79-fold. Suppressed P-gp efflux function. Inhibition of ERK1/2 and AKT phosphorylation. |
| MCF-7/ADR | At 5 µM reduced the GI50 1 of vincristine 151.04-fold and paclitaxel 151.07-fold. | ||||
| KB-C2 cell xenografts in nude mice model | At 15 mg/kg significantly enhanced the anticancer activity of paclitaxel (18 mg/kg), with no significant change in the body weight (Reduced toxicity). | ||||
| 113 | OH | OH |  | HepG2/ADM | At 4 µM reduced the GI50 1 of vincristine 48.03–fold and paclitaxel 82-fold. Inhibited P-gp ATP-ase activity and suppress its efflux function |
| MCF-7/ADR | At 4 µM reduced the GI50 1 of vincristine 29-fold and paclitaxel 47-fold. | ||||
| 114 | OAc | OAc | | HepG2/ADM | At 4 µM reduced the GI50 1 of vincristine 232-fold and paclitaxel 282-fold. Inhibited P-gp ATP-ase activity and suppress its efflux function. |
| MCF-7/ADR | At 4 µM reduced the GI50 1 of vincristine 183-fold and paclitaxel 59-fold. | ||||
| verapamil (1)2 | - | - | - | HepG2/ADM | At 5 µM reduced the GI50 1 of vincristine 22-fold and paclitaxel 11-fold. |
| MCF-7/ADR | At 5 µM reduced the GI50 1 of vincristine 29-fold and paclitaxel 59-fold. |
| Compound | ABC Protein Target | Cell Line/Animal Model | Activity/Mechanism of Action | Ref. |
|---|---|---|---|---|
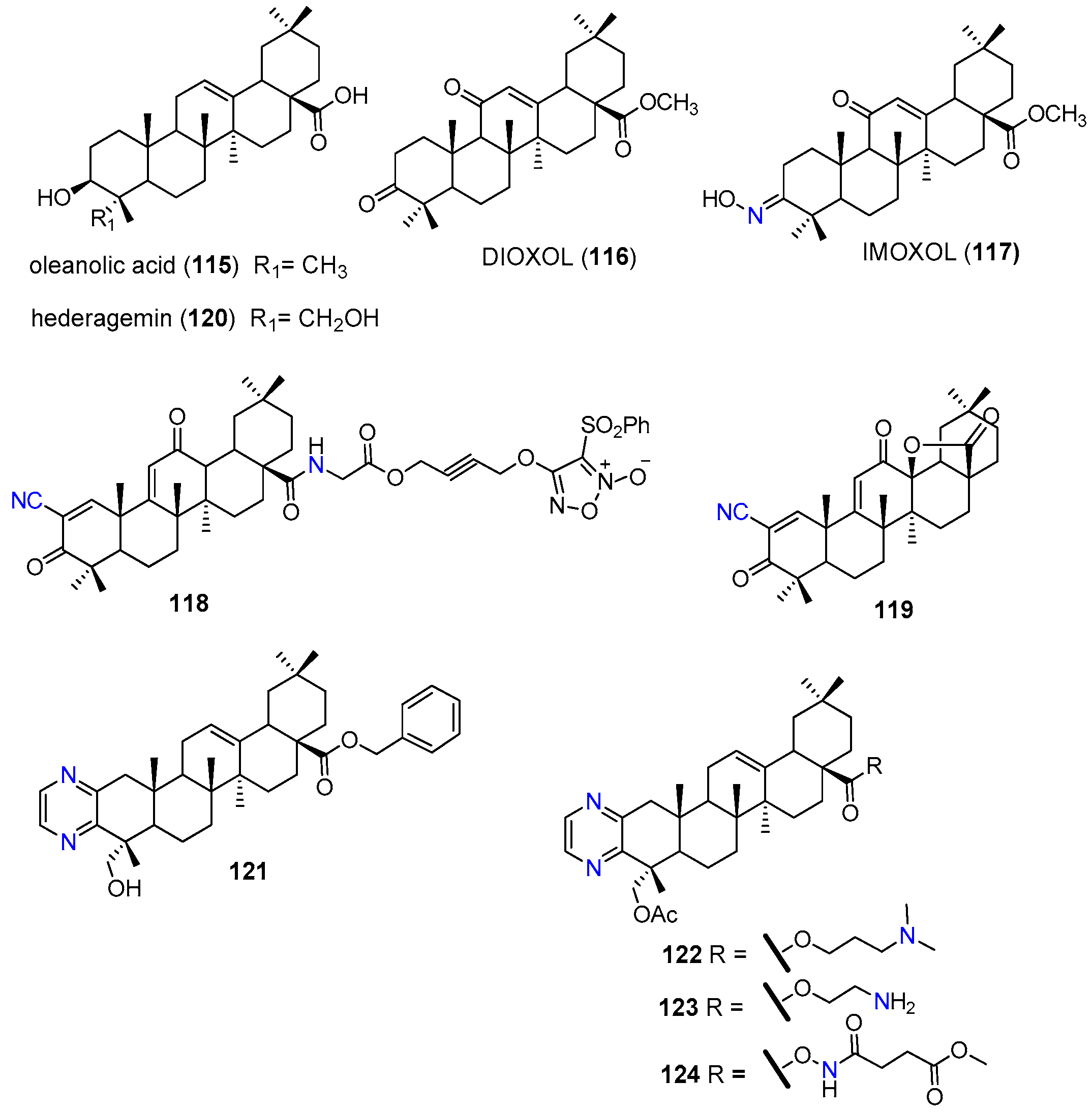
| 117 | MRP1 | HL-60/AR | Inhibited MRP1 transport function (short term response). Reduced the mNMR and protein expression levels of MRP1. | [135] |
| 118 | P-gp, MRP1 BCRP | HCT-8/5-FU | Reduced the relative levels of P-gp, MRP1, and BCRP by nitrating these cellular drug efflux proteins. | [136] |
| 119 | P-gp | A549/CDDP | Reduced the P-gp expression at the protein and mRNA levels. Suppressed P-gp ATPase activity. Inhibited MEK/ERK and PI3K/AKT pathways. Inhibited TrxR expression and activity. | [137] |
| 121 | P-gp | KBV | At 10 µM reduced the GI50 1 of paclitaxel 158-fold and the GI50 1 of vincristine 78-fold. Inhibited the P-gp efflux activity without affecting its protein expression. Increased P-gp ATP-ase activity. | [138] |
| MCF7/T | At 10 µM reduced the GI50 1 of paclitaxel 14-fold and the IC50 of vincristine 33-fold. | |||
| KBV xenograft in nude mice | At 10 mg/kg significantly enhanced anti-tumor activity of paclitaxel with good safety profile. | |||
| 122 | P-gp | KBV | At 10 µM reduced the GI50 1 of paclitaxel 80.6-fold Increased P-gp ATPase activity. | [139] |
| KBV xenograft in nude mice | Enhanced antitumor activity of paclitaxel. |
| Compound | Cells line/Animal Model | Activity/Mechanism of Action | Ref. |
|---|---|---|---|
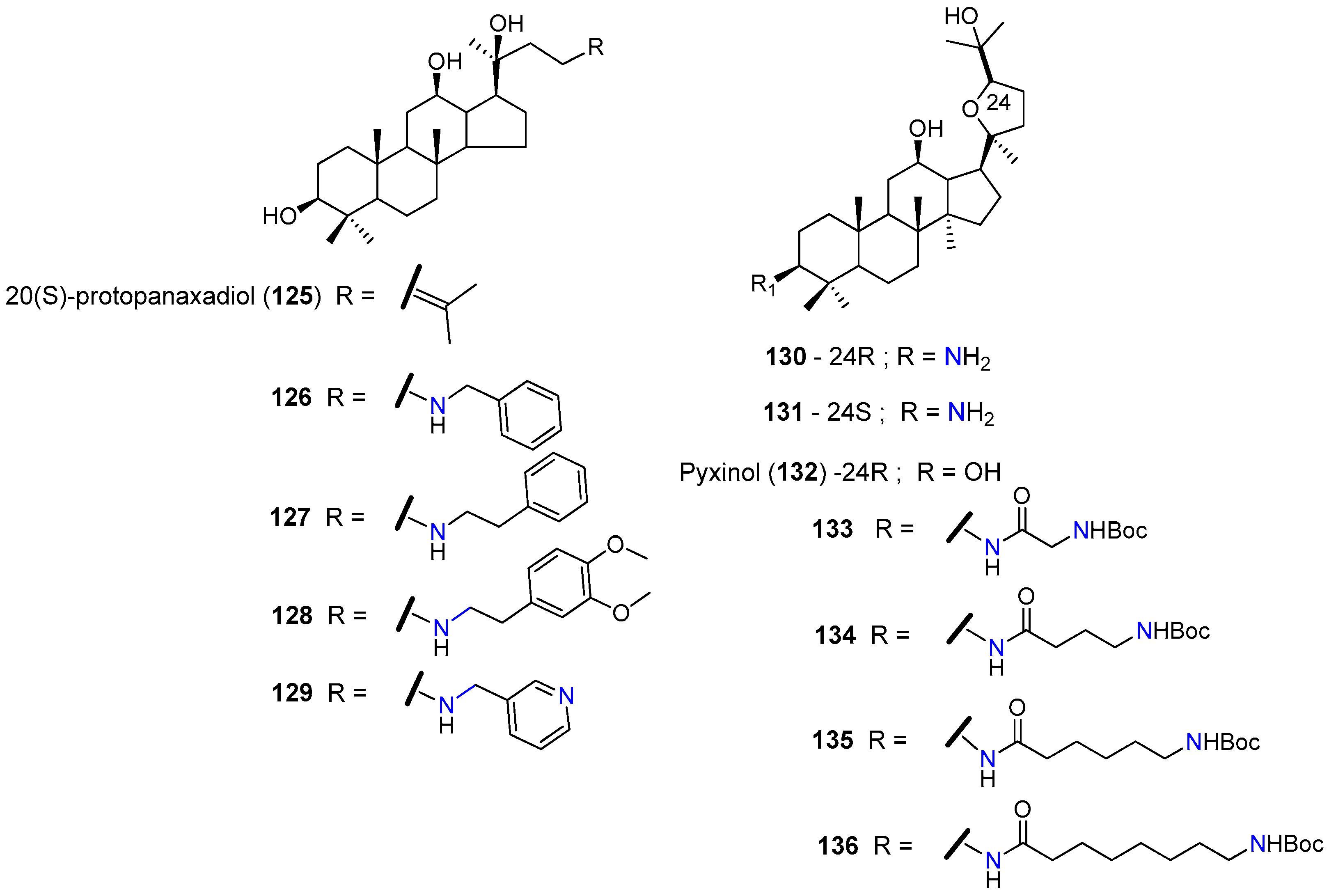
| 126 | KBvcr cells | Sensitized multidrug-resistant cells to docetaxel, vincristine and doxorubicin. At 5 µM reduced the GI50 1 of docetaxel 111-fold, the GI50 of vincristine 87-fold, and the GI50 of doxorubicin 56-fold. EC50 2 = 1.367 µM for doxorubicin resistance. Inhibited P-gp–efflux function. | [144] |
| KBvcr xenografts mice model | At 100 mg/kg significantly enhanced the inhibitory effect of doxorubicin against a multidrug-resistant tumor in the xenograft model. | [145,146] | |
| 127 | KBvcr cells | Sensitized multidrug-resistant cells to docetaxel, vincristine and doxorubicin. At 5 µM reduced the GI50 1 of doxorubicin 196-fold, the GI50 1 of vincristine 117-fold, and the GI50a of Adriamycin 62-fold. EC50 2 = 1.31 µM for doxorubicin resistance. | [144] |
| 130 | SW620/Ad300 | At 3 µM significantly reversed the resistance to paclitaxel and vincristine by reducing the GI50 1 of paclitaxel 26-fold and the GI50 1 of vincristine 28-fold. Suppressed the efflux function of P-gp. Stimulated the ATPase activity of P-gp. | [147] |
| HEK/ABCB1 | At 3 µM reduced the GI50a of paclitaxel 13.00-fold and the GI50 1 of vincristine 5.58-fold. | ||
| 131 | SW620/Ad300 | At 3 µM reduced the GI50 1 of paclitaxel 1.41-fold and the GI50 1 of vincristine 1.81-fold. | [147] |
| 135 | KBV | At 5 µM reduced the GI50 1 of paclitaxel 28-fold. Significantly increased the population of paclitaxel-treated cells in the G2-M phase. Increased the accumulation of Rhodamine123 in KBV cells in a dose-dependent manner. Increased the P-gp-ATPase activity over the basal level by 6.03-fold. | [148] |
| KBV xenograft nude mice model | Oral administration of 10 mg/kg, significantly increased the tumor inhibitory activity of paclitaxel (30 mg/kg). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. F. Gonçalves, B.; S. P. Cardoso, D.; U. Ferreira, M.-J. Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators. Molecules 2020, 25, 3364. https://doi.org/10.3390/molecules25153364
M. F. Gonçalves B, S. P. Cardoso D, U. Ferreira M-J. Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators. Molecules. 2020; 25(15):3364. https://doi.org/10.3390/molecules25153364
Chicago/Turabian StyleM. F. Gonçalves, Bruno, David S. P. Cardoso, and Maria-José U. Ferreira. 2020. "Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators" Molecules 25, no. 15: 3364. https://doi.org/10.3390/molecules25153364
APA StyleM. F. Gonçalves, B., S. P. Cardoso, D., & U. Ferreira, M. -J. (2020). Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators. Molecules, 25(15), 3364. https://doi.org/10.3390/molecules25153364