Ab Initio Molecular Dynamics Study of Methanol-Water Mixtures under External Electric Fields




Abstract
:1. Introduction
2. Materials and Methods
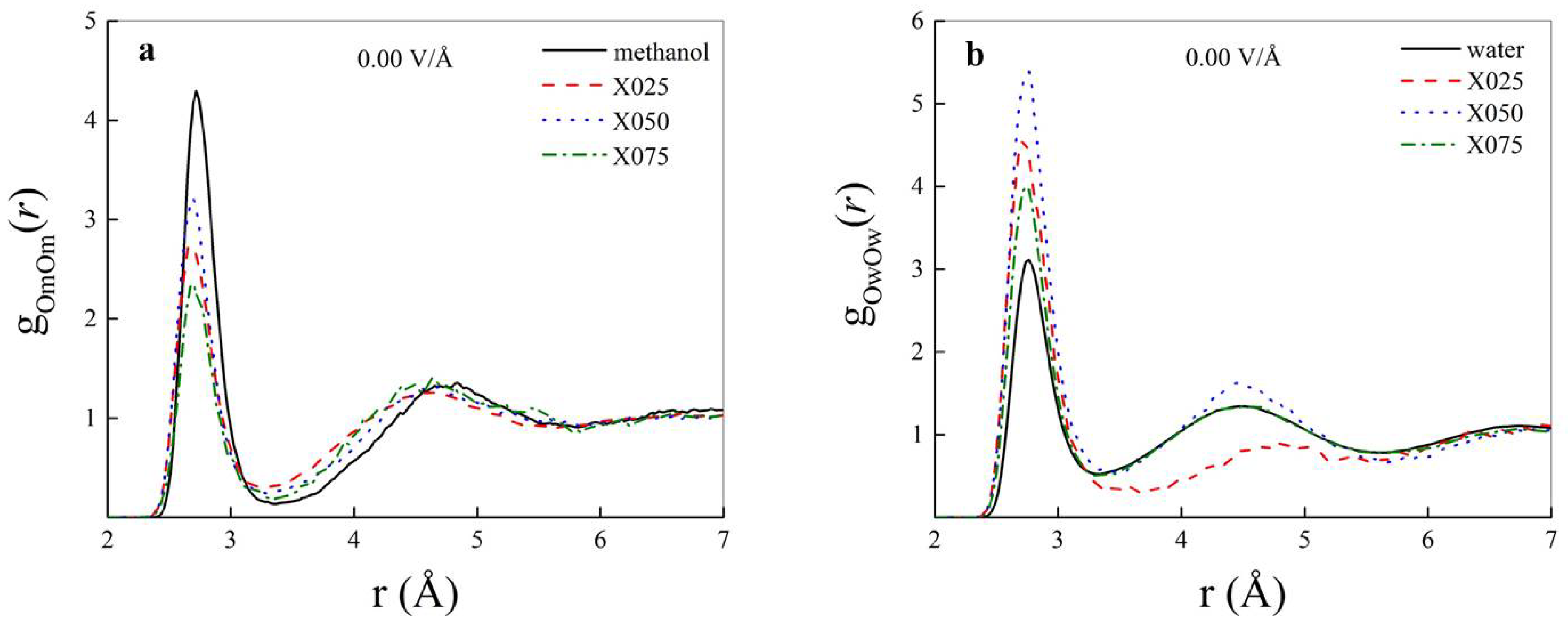
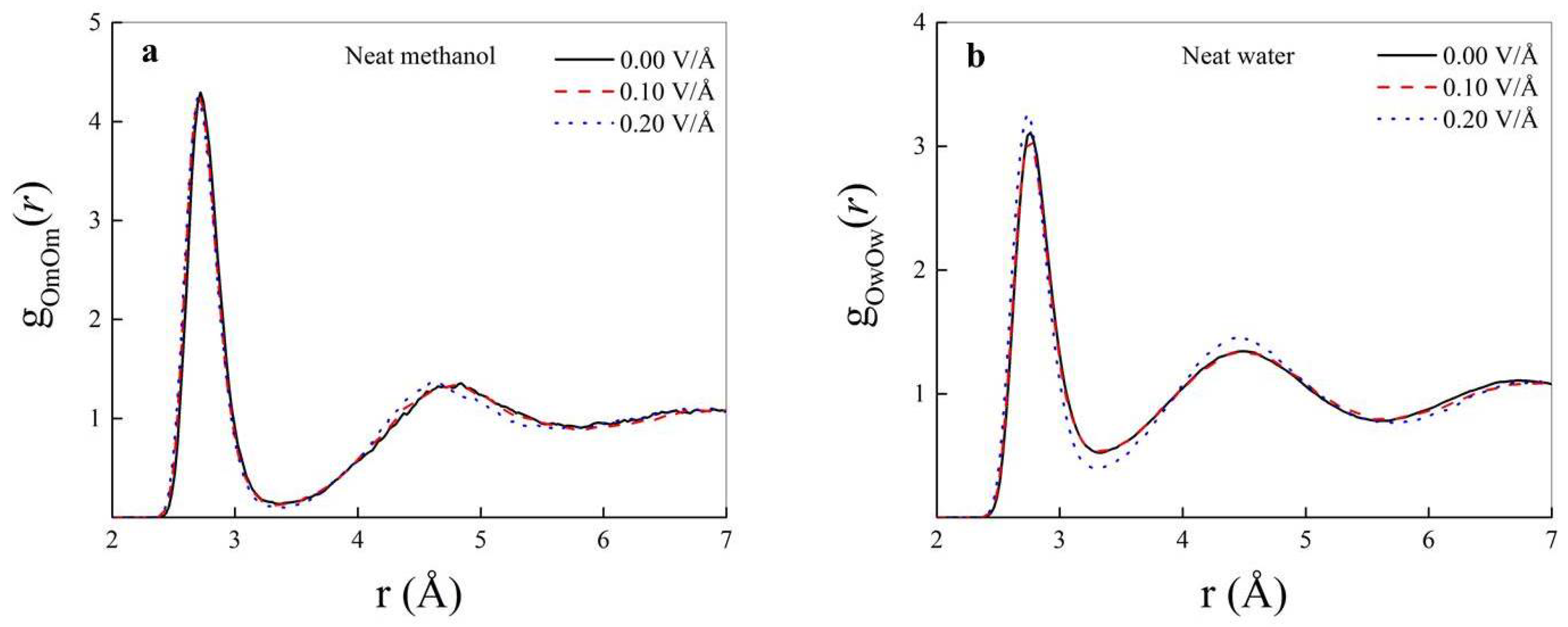
3. Results and Discussion
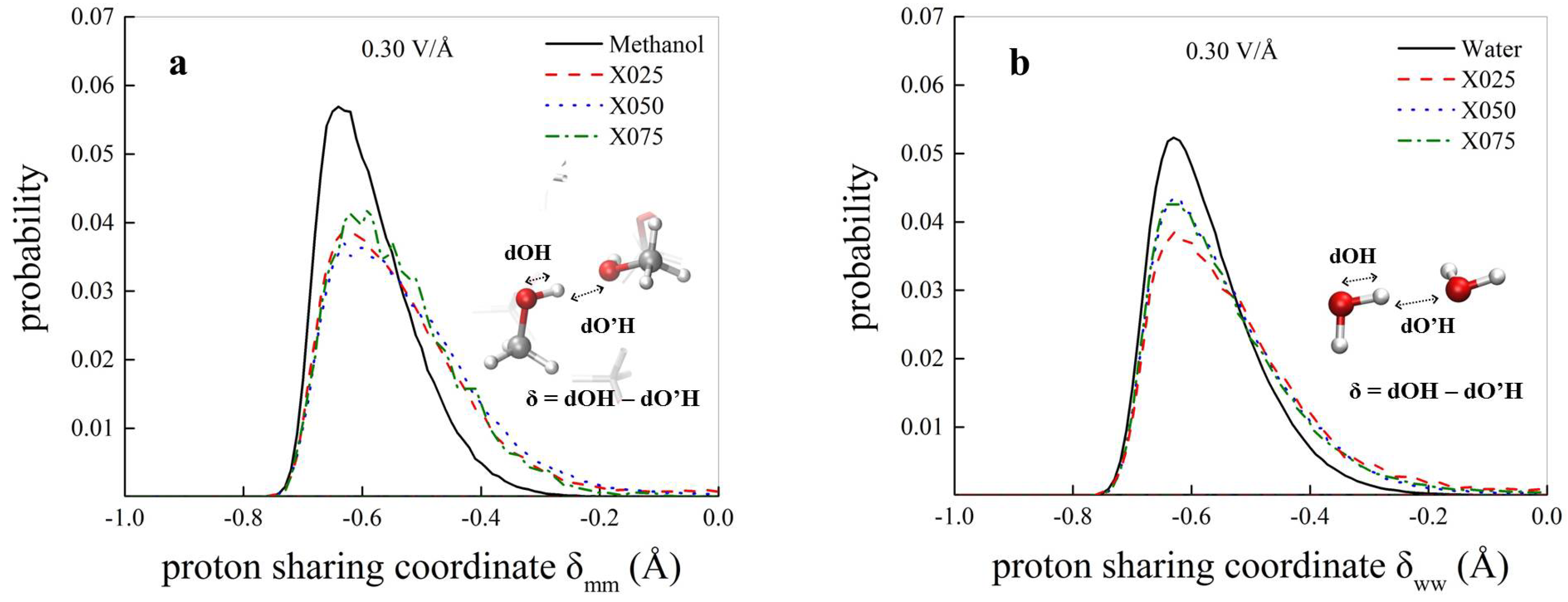
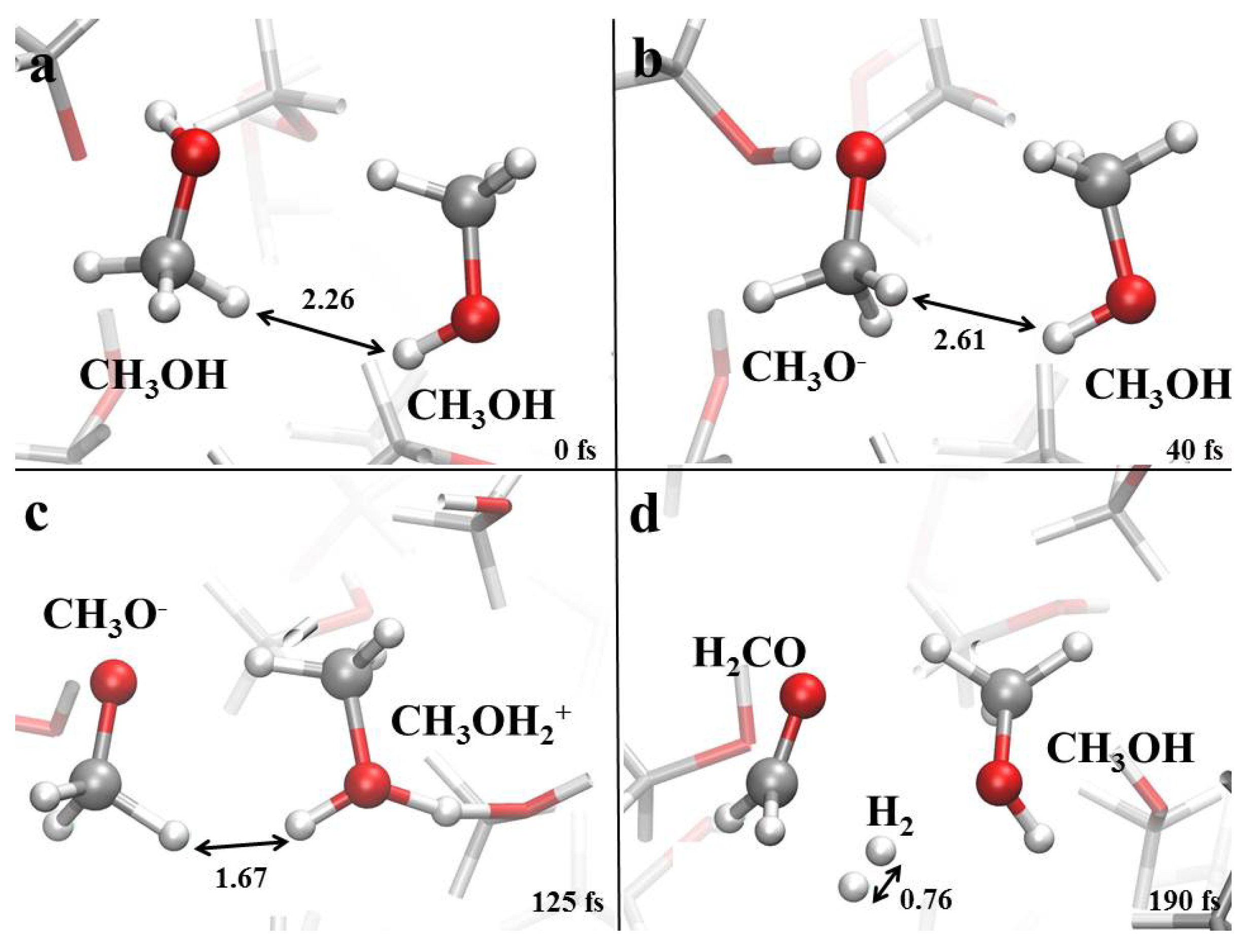
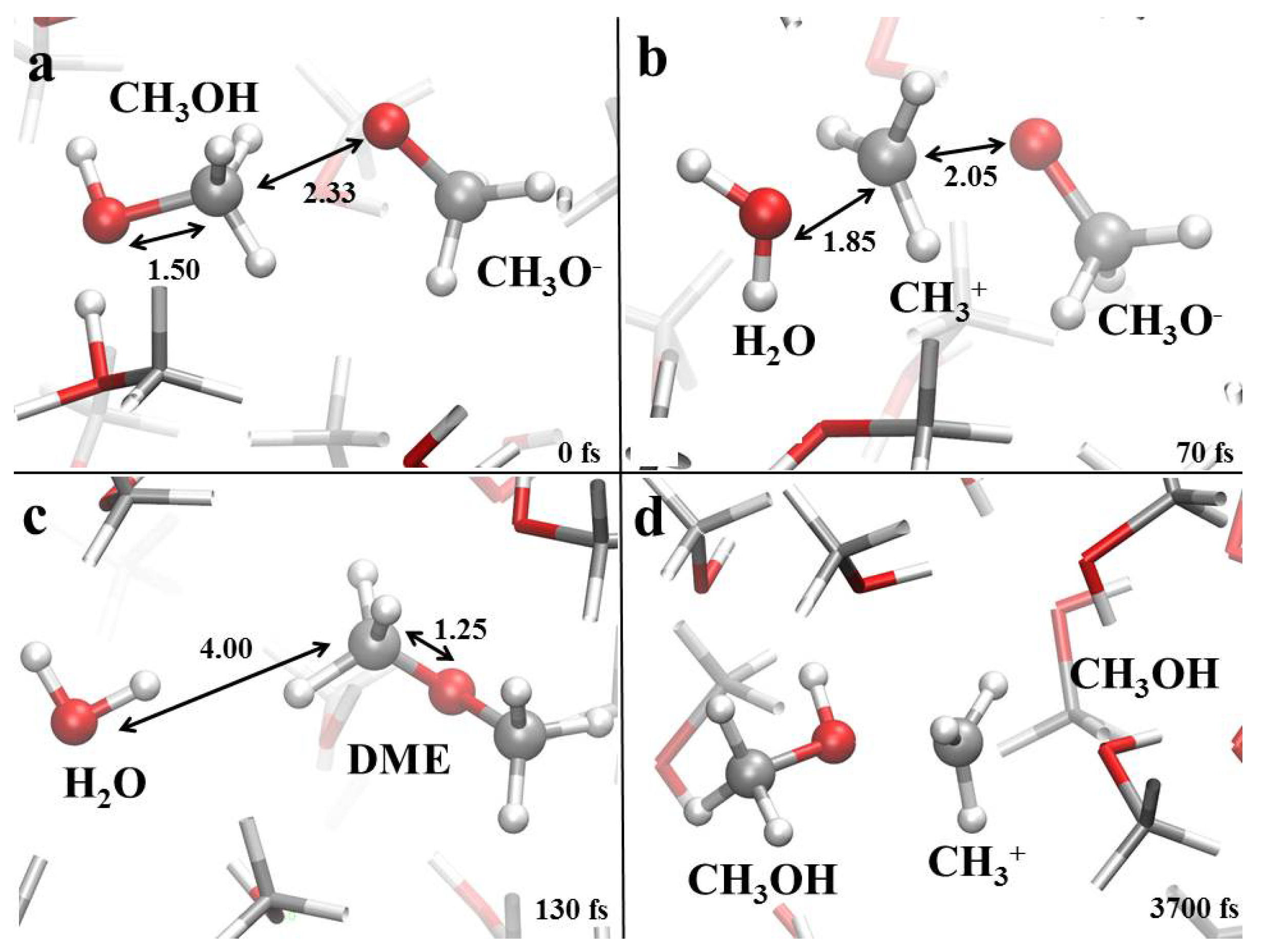
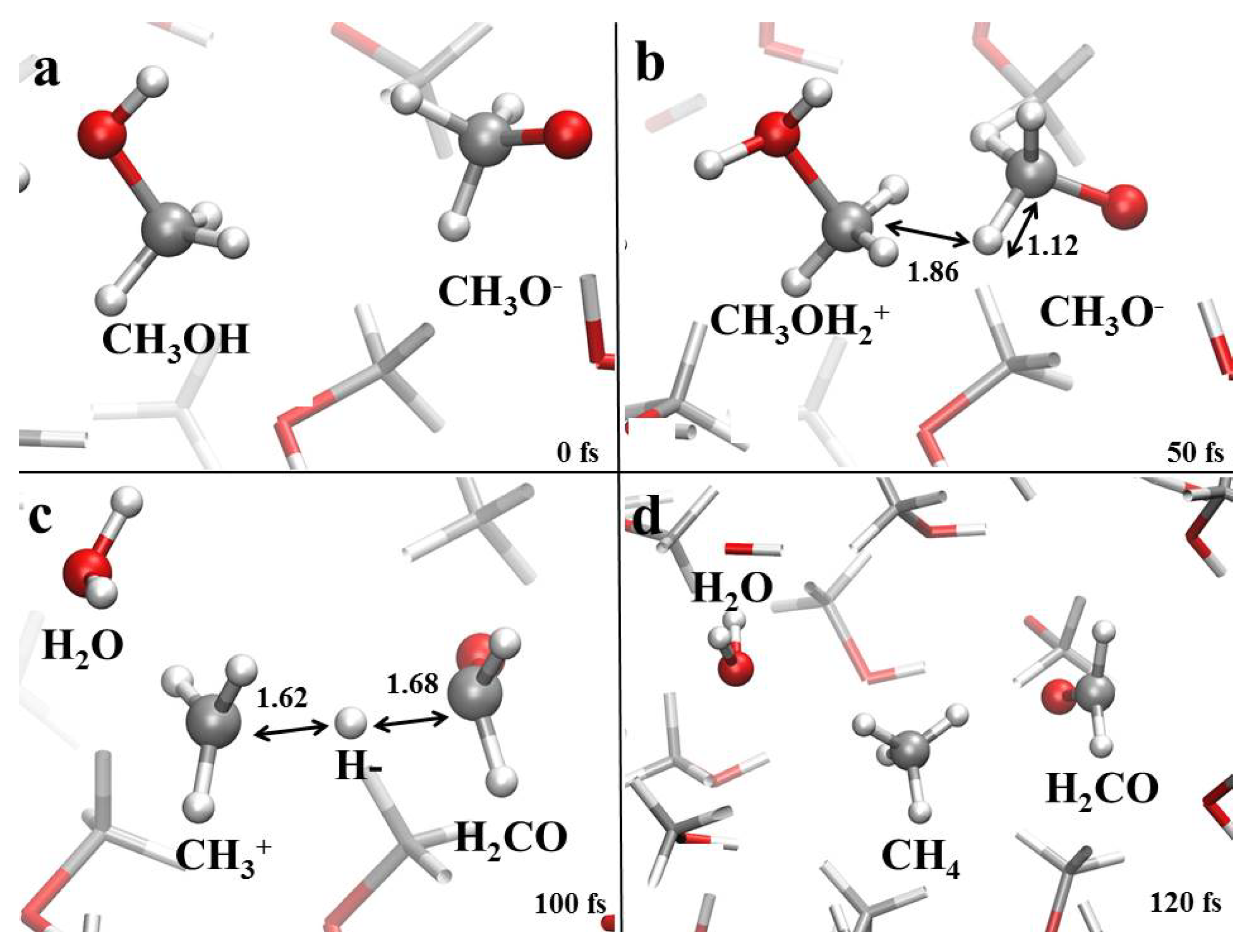
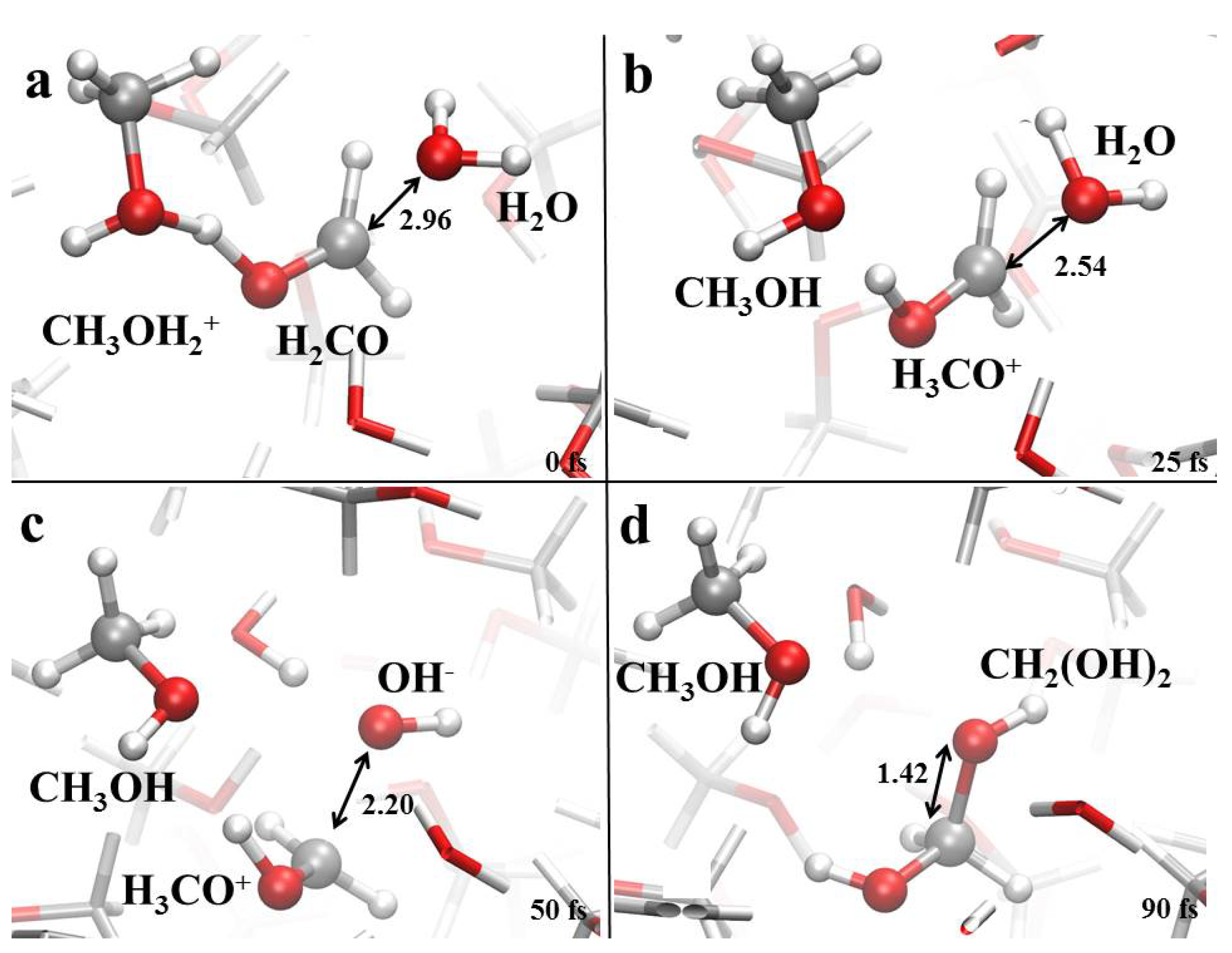
3.1. Field-Induced Proton Transfer
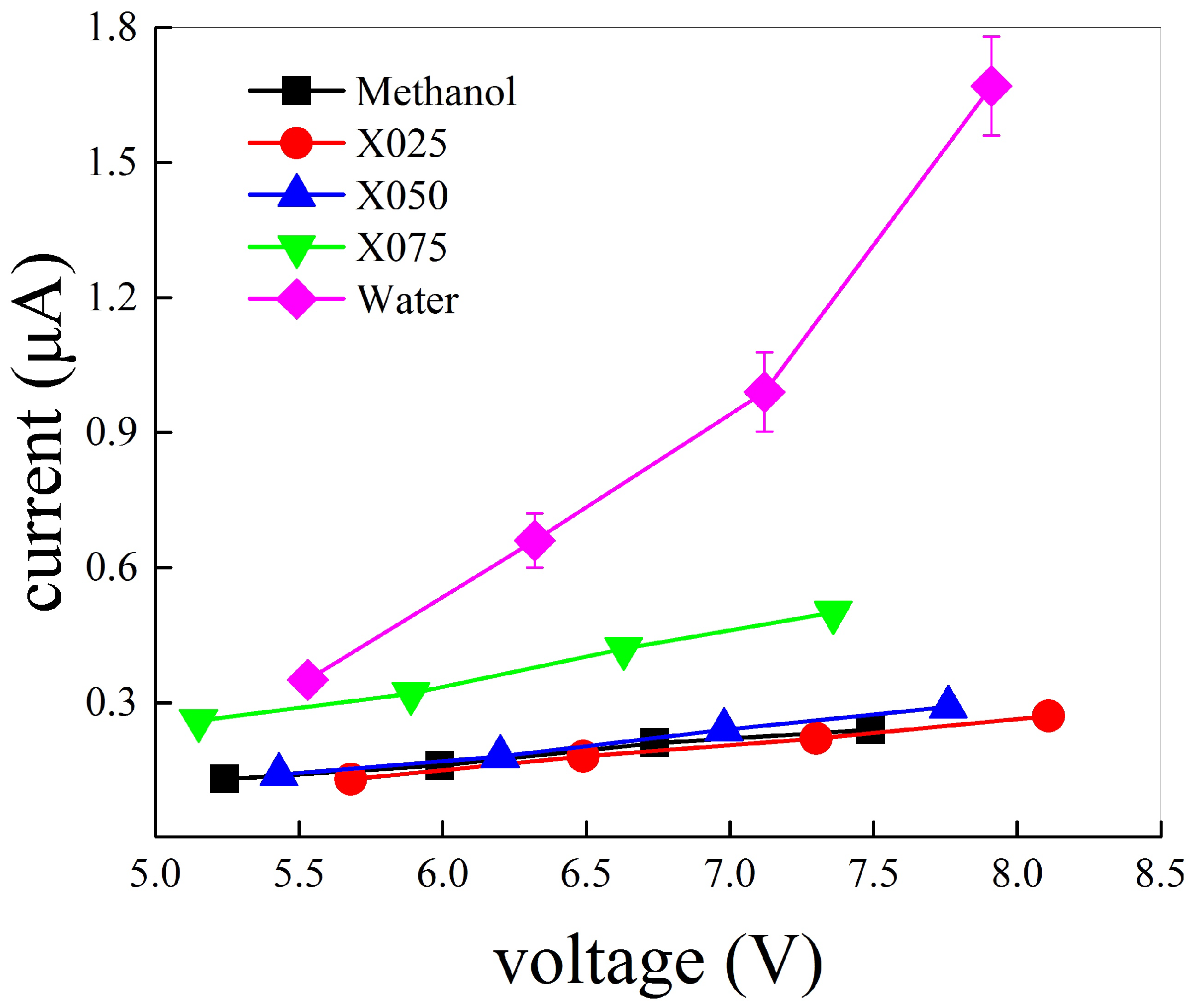
3.2. Field-Induced Chemical Reactions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Franks, F.; Reid, D.S. Water-A Comprehensive Treatise; Franks, F., Ed.; Plenum: New York, NY, USA, 1973; Volume 2, Chapter 5. [Google Scholar]
- Pratt, J.R.; Chandler, D. Effects of solute–solvent attractive forces on hydrophobic correlations. J. Chem. Phys. 1980, 73, 1980. [Google Scholar] [CrossRef]
- Gurav, N.D.; Kulkarni, A.D.; Gejji, S.P.; Pathak, R.K. CH3OH (H2O)n [n = 1–4] clusters in external electric fields. J. Chem. Phys. 2015, 142, 214309. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Crain, J.; Poon, W.C.K.; Finney, J.L.; Soper, A.K. Molecular segregation observed in a concentrated alcohol–water solution. Nature 2002, 416, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.H.; Luo, Y.; Augustsson, A.; Kashtonov, S.; Rubensson, J.E.; Shuh, D.H.; Agren, H.; Norgren, J. Molecular Structure of Alcohol-Water Mixtures. Phys. Rev. Lett. 2003, 91, 157401. [Google Scholar] [CrossRef]
- Dougan, L.; Bates, S.P.; Hargreaves, R.; Fox, J.P.; Crain, J.; Finney, J.L.; Reat, V.; Soper, A.K. Methanol-water solutions: A bi-percolating liquid mixture. J. Chem. Phys. 2004, 121, 6456. [Google Scholar] [CrossRef] [Green Version]
- Lenton, S.; Rhys, N.H.; Towey, J.J.; Soper, A.K.; Dougan, L. Temperature-Dependent Segregation in Alcohol-Water Binary Mixtures Is Driven by Water Clustering. J. Phys. Chem. B 2018, 122, 7884–7894. [Google Scholar] [CrossRef]
- Mallamace, F.; Corsaro, C.; Mallamace, D.; Vasi, C.; Vasi, S.; Stanley, H.E. Dynamical properties of water-methanol solutions. J. Chem. Phys. 2016, 144, 064506. [Google Scholar] [CrossRef] [Green Version]
- Nakasaga, M.; Mochizuki, K.; Leloup, V.; Kosugi, N. Local Structures of Methanol-Water Binary Solutions Studied by Soft X-ray Absorption Spectroscopy. J. Phys. Chem. B 2014, 118, 4388–4396. [Google Scholar] [CrossRef]
- Galicia-Andres, E.; Pusztai, L.; Temleitner, L.; Pizio, O. Microscopic structure of methanol–water mixtures: Synchrotron X-ray diffraction experiments and molecular dynamics simulations over the entire composition range. J. Mol. Liq. 2015, 209, 586–595. [Google Scholar] [CrossRef]
- Sato, T.; Chiba, A.; Ryusuke, N. Hydrophobic hydration and molecular association in methanol-water mixtures studied by microwave dielectric analysis. J. Chem. Phys. 2000, 112, 2924. [Google Scholar] [CrossRef]
- Vezzu’, K.; Negro, E.; He, J.; Bertasi, F.; Conti, F.; Nawn, G.; Paddison, S.J.; Di Noto, V. Reorientational Relaxation and Hydrogen Bonding in Mixtures of Water and Methanol. J. Electrochem. Soc. 2018, 165, H549–H560. [Google Scholar] [CrossRef]
- Ferrario, M.; Haughney, M.; McDonald, I.R.; Klein, M.L. Molecular-dynamics simulation of aqueous mixtures: Methanol, acetone, and ammonia. J. Chem. Phys. 1990, 93, 5156. [Google Scholar] [CrossRef]
- Tanaka, H.; Gubbins, K.E. Structure and thermodynamic properties of water-methanol mixtures: Role of the water-water interaction. J. Chem. Phys. 1998, 97, 2626. [Google Scholar] [CrossRef]
- Laaksonen, A.; Kusalik, P.G.; Svishchev, I.M. Three-Dimensional Structure in Water-Methanol Mixtures. J. Phys. Chem. A 1997, 101, 5910–5918. [Google Scholar] [CrossRef]
- Wensink, E.J.W.; Hoffmann, A.C.; van Maaren, P.J.; van der Spoel, D. Dynamic properties of water-alcohol mixtures studied by computer simulation. J. Chem. Phys. 2003, 119, 7308. [Google Scholar] [CrossRef] [Green Version]
- Bako, C.I.; Megyes, T.; Balint, S.; Grosz, T.; Chihaia, V. Water-methanol mixtures: Topology of hydrogen bonded network. Phys. Chem. Chem. Phys. 2008, 10, 5004–5011. [Google Scholar] [CrossRef]
- Zhang, N.; Shen, Z.; Chen, C.; He, G.; Hao, C. Effect of hydrogen bonding on self-diffusion in methanol/water liquid mixtures: A molecular dynamics simulation study. J. Mol. Phys. 2015, 203, 90–97. [Google Scholar] [CrossRef] [Green Version]
- galicia-andres, E.; Dominguez, A.; Pusztai, L.; Pizio, O. On the composition dependence of thermodynamic, dynamic and dielectric properties of water-methanol model mixtures. Molecular dynamics simulation results. Condens. Matter. Phys. 2015, 18, 43602. [Google Scholar] [CrossRef] [Green Version]
- Soetens, J.C.; Bopp, P.A. Water-Methanol Mixtures: Simulations of Mixing Properties over the Entire Range of Mole Fractions. J. Phys. Chem. B 2015, 119, 8593–8599. [Google Scholar] [CrossRef]
- Alberti, M.; Amat, A.; Aguilar, A.; Pirani, F. Methanol–methanol and methanol-water systems: The intermolecular interactions controlling the transition from small clusters to the liquid phase. Phys. Chem. Chem. Phys. 2017, 19, 16765–16774. [Google Scholar] [CrossRef]
- Kacar, G.; de With, G. Parametrizing hydrogen bond interactions in dissipative particle dynamics simulations: The case of water, methanol and their binary mixtures. J. Mol. Phys. 2020, 302, 112581. [Google Scholar] [CrossRef]
- van Erp, T.S.; Meijer, E.J. Hydration of methanol in water. A DFT-based molecular dynamics study. Chem. Phys. Lett. 2001, 333, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Silvestrelli, P.G. Are There Immobilized Water Molecules around Hydrophobic Groups? Aqueous Solvation of Methanol from First Principles. J. Phys. Chem. B 2009, 113, 10728–10731. [Google Scholar] [CrossRef] [PubMed]
- Chouduri, J.R.; Chandra, A. Hydrogen bonded structure, polarity, molecular motion and frequency fluctuations at liquid-vapor interface of a water-methanol mixture: An ab initio molecular dynamics study. J. Chem. Phys. 2014, 141, 134703. [Google Scholar] [CrossRef] [PubMed]
- Morrone, J.A.; Haslinger, K.E.; Tuckerman, M.E. Ab Initio Molecular Dynamics Simulation of the Structure and Proton Transport Dynamics of Methanol-Water Solutions. J. Phys. Chem. B 2006, 110, 3712–3720. [Google Scholar] [CrossRef]
- Suresh, S.J. Influence of electric field on the hydrogen bond network of methanol. J. Chem. Phys. 2007, 126, 134502. [Google Scholar] [CrossRef]
- Cassone, G.; Giaquinta, P.V.; Saija, F.; Saitta, A.M. Liquid methanol under a static electric field. J. Chem. Phys. 2015, 142, 054502. [Google Scholar] [CrossRef]
- Saitta, A.M.; Saija, F.; Giaquinta, P.V. Ab Initio Molecular Dynamics Study of Dissociation of Water under an Electric Field. Phys. Rev. Lett. 2012, 108, 207801. [Google Scholar] [CrossRef] [Green Version]
- Cassone, G.; Giaquinta, P.V.; Saija, F.; Saitta, A.M. Proton Conduction in Water Ices under an Electric Field. J. Phys. Chem. B 2014, 118, 4419–4424. [Google Scholar] [CrossRef]
- Shafiei, M.; von Domaros, M.; Bratko, D.; Luzar, A. Anisotropic structure and dynamics of water under static electric fields. J. Chem. Phys. 2019, 150, 074505. [Google Scholar] [CrossRef]
- Cassone, G.; Sponer, J.; Trusso, S.; Saija, F. Ab initio spectroscopy of water under electric fields. Phys. Chem. Chem. Phys. 2019, 21, 21205–21212. [Google Scholar] [CrossRef] [PubMed]
- Aragones, A.C.; Haworth, N.L.; Darwish, N.; Ciampi, S.; Bloomfield, G.J.; Wallace, G.G.; Diez-Perez, I.; Coote, M.L. Electrostatic catalysis of a Diels-Alder reaction. Nature 2016, 531, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Mandal, D.; Ramanan, R. Oriented electric fields as future smart reagents in chemistry. Nat. Chem. 2016, 8, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Che, F.; Gray, J.T.; Ha, S.; Kruse, N.; Scott, S.L.; McEwen, J.-S. Elucidating the Roles of Electric Fields in Catalysis: A Perspective. ACS Catal. 2018, 8, 5153–5174. [Google Scholar] [CrossRef]
- Cassone, G.; Pietrucci, F.; Saija, F.; Guyot, F.; Saitta, A.M. One-step electric-field driven methane and formaldehyde synthesis from liquid methanol. Chem. Sci. 2017, 8, 2329–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassone, G.; Pietrucci, F.; Saija, F.; Guyot, F.; Sponer, J.E.; Sponer, J.; Saitta, A.M. Novel electrochemical route to cleaner fuel dimethyl ether. Sci. Rep. 2017, 7, 6901. [Google Scholar] [CrossRef] [Green Version]
- Kaila, K.; Ranson, B.R. pH and Brain Function; Kaila, K., Ransom, B.R., Eds.; Wiley: New York, NY, USA, 1998. [Google Scholar]
- He, J.; Di Noto, V.; Paddison, S.J. The structure of water-methanol mixtures under an electric field: Ab initio molecular dynamics simulations. Chem. Phys. Lett. 2015, 635, 99–106. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2014, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate Density Functional calculations using a mixed gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103. [Google Scholar] [CrossRef] [Green Version]
- King-Smith, R.D.; Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B. 1993, 47, 1651. [Google Scholar] [CrossRef]
- Resta, R. Macroscopic polarization in crystalline dielectrics: The geometric phase approach. Rev. Mod. Phys. 1994, 66, 899. [Google Scholar] [CrossRef]
- Berry, M.V. Quantal phase factors accompanying adiabatic changes. Proc. R. Soc. Lond. A 1984, 392, 45. [Google Scholar]
- Umari, P.; Pasquarello, A. Ab initio molecular dynamics in a finite homogeneous electric field. Phys. Rev. Lett. 2002, 89, 157602. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; Waldron, J.C. Perspectives on external electric fields in molecular simulation: Progress, prospects and challenges. Phys. Chem. Chem. Phys. 2015, 17, 12407–12440. [Google Scholar] [CrossRef] [PubMed]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of Density Functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected Density Functional Theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Lin, I.-C.; Seitsonen, A.P.; Tavernelli, I.; Rothlisberger, U. Structure and dynamics of liquid water from ab initio molecular dynamics—comparison of BLYP, PBE, and revPBE Density Functionals with and without van der Waals corrections. J. Chem. Theory Comput. 2012, 8, 3902–3910. [Google Scholar] [CrossRef]
- Bankura, A.; Karmakar, A.; Carnevale, V.; Chandra, A.; Klein, M.L. Structure, dynamics, and spectral diffusion of water from first-principles molecular dynamics. J. Phys. Chem. C 2014, 118, 29401–29411. [Google Scholar] [CrossRef]
- Gillan, M.J.; Alfé, D.; Michaelides, A. Perspective: How good is DFT for water? J. Chem. Phys. 2016, 144, 130901. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Hidaka, K.; Soper, A.K. The structure of liquid methanol revisited: A neutron diffraction experiment at −80 °C and +25 °C. Mol. Phys. 1999, 96, 1159–1168. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hidaka, K.; Soper, A.K. The structure of liquid methanol revisited: A neutron diffraction experiment at −80 °C and +25 °C. Mol. Phys. 1999, 97, 603–605. [Google Scholar] [CrossRef]
- Cassone, G.; Sofia, A.; Rinaldi, G.; Sponer, J. Catalyst-Free Hydrogen Synthesis from Liquid Ethanol: An ab Initio Molecular Dynamics Study. J. Phys. Chem. C 2019, 123, 9202–9208. [Google Scholar] [CrossRef] [Green Version]
- Vanzo, D.; Bratko, D.; Luzar, A. Nanoconfined water under electric field at constant chemical potential undergoes electrostriction. J. Chem. Phys. 2014, 140, 074710. [Google Scholar] [CrossRef]
- Stuve, E.M. Ionization of water in interfacial electric fields: An electrochemical view. Chem. Phys. Lett. 2012, 519–520, 1–17. [Google Scholar] [CrossRef]
- Lee, W.K.; Tsoi, S.; Whitener, K.E.; Stine, R.; Robinson, J.T.; Tobin, J.S.; Weerasinghe, A.; Sheehan, P.E.; Lyuksyutov, S.F. Robust reduction of graphene fluoride using an electrostatically biased scanning probe. Nano Res. 2013, 6, 767–774. [Google Scholar] [CrossRef]
- Hammadi, Z.; Descoins, M.; Salançon, E.; Morin, R. Proton and light ion nanobeams from field ionization of water. Appl. Phys. Lett. 2012, 101, 243110. [Google Scholar] [CrossRef]
- Ceriotti, M.; Cuny, J.; Parrinello, M.; Manolopoulos, D.E. Nuclear quantum effects and hydrogen bond fluctuations in water. Proc. Natl. Acad. Sci. USA 2013, 110, 15591–15596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsalek, O.; Markland, T.E. Quantum dynamics and spectroscopy of ab initio liquid water: The interplay of nuclear and electronic quantum effects. J. Phys. Chem. Lett. 2017, 8, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Gaiduk, A.P.; Gygi, F.; Galli, G. Density and compressibility of liquid water and ice from first-principles simulations with hybrid functionals. J. Phys. Chem. Lett. 2015, 6, 2902–2908. [Google Scholar] [CrossRef] [PubMed]
- Miceli, G.; de Gironcoli, S.; Pasquarello, A. Isobaric first-principles molecular dynamics of liquid water with nonlocal van der Waals interactions. J. Chem. Phys. 2015, 142, 034501. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zheng, L.; Santra, B.; Ko, H.-Y.; DiStasio, R.A., Jr.; Klein, M.L.; Car, R.; Wu, X. Hydroxide diffuses slower than hydronium in water because its solvated structure inhibits correlated proton transfer. Nat. Chem. 2018, 10, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.D.; Boxer, S.G. Electric Fields and Enzyme Catalysis. Annu. Rev. Biochem. 2017, 86, 387–415. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Electron-Transfer Processes of Cytochrome c at Interfaces. New Insights by Surface-Enhanced Resonance Raman Spectroscopy. Acc. Chem. Res. 2004, 37, 854–861. [Google Scholar] [CrossRef]
- Saitta, A.M.; Saija, F. Miller experiments in atomistic computer simulations. Proc. Natl. Acad. Sci. USA 2014, 111, 13768–13773. [Google Scholar] [CrossRef] [Green Version]
- Sowlati-Hashjin, S.; Matta, C.F. The chemical bond in external electric fields: Energies, geometries, and vibrational Stark shifts of diatomic molecules. J. Chem. Phys. 2013, 139, 144101. [Google Scholar] [CrossRef] [Green Version]
- Papanikolaou, P.; Karafiloglou, P. Investigating sigma bonds in an electric field from the Pauling’s perspective: The behavior of Cl-X and H-X (X = C,Si) bonds. Theor. Chem. Acc. 2010, 126, 213–222. [Google Scholar] [CrossRef]
- Rincon, L.; Mora, J.R.; Torres, F.J.; Almeida, R. On the activation of σ-bonds by electric fields: A Valence Bond perspective. Chem. Phys. 2016, 477, 1–7. [Google Scholar] [CrossRef]
- Stark, J. Observation of the Separation of Spectral Lines by an Electric Field. Nature 1913, 92, 401. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.M. The vibrational Stark effect. J. Chem. Phys. 1993, 98, 3179. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Boxer, S.G. Vibrational Stark Effect Spectroscopy. J. Am. Chem. Soc. 1995, 117, 1449–1450. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Species (%) | Methanol | 75:25 mix | 50:50 mix | 25:75 mix | Water |
|---|---|---|---|---|---|
| Formaldehyde | 4 | 2 | − | − | − |
| Methane | 2 | 2 | − | − | − |
| Hydrogen | 2 | 2 | − | − | − |
| Water | 6 | 5 | 3 | − | − |
| Formaldehyde monohydrate | − | 2 | − | − | − |
| Dimethyl ether (DME) | 6 | 2 | 3 | − | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassone, G.; Sofia, A.; Sponer, J.; Saitta, A.M.; Saija, F. Ab Initio Molecular Dynamics Study of Methanol-Water Mixtures under External Electric Fields. Molecules 2020, 25, 3371. https://doi.org/10.3390/molecules25153371
Cassone G, Sofia A, Sponer J, Saitta AM, Saija F. Ab Initio Molecular Dynamics Study of Methanol-Water Mixtures under External Electric Fields. Molecules. 2020; 25(15):3371. https://doi.org/10.3390/molecules25153371
Chicago/Turabian StyleCassone, Giuseppe, Adriano Sofia, Jiri Sponer, A. Marco Saitta, and Franz Saija. 2020. "Ab Initio Molecular Dynamics Study of Methanol-Water Mixtures under External Electric Fields" Molecules 25, no. 15: 3371. https://doi.org/10.3390/molecules25153371
APA StyleCassone, G., Sofia, A., Sponer, J., Saitta, A. M., & Saija, F. (2020). Ab Initio Molecular Dynamics Study of Methanol-Water Mixtures under External Electric Fields. Molecules, 25(15), 3371. https://doi.org/10.3390/molecules25153371