
A Pilot Study of Multi-Input Recurrent Neural Networks for Drug-Kinase Binding Prediction



Abstract
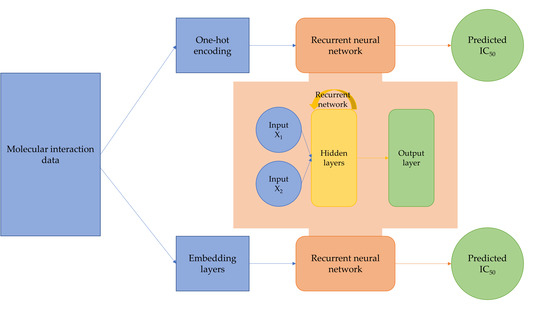
:
1. Introduction
2. Results
2.1. Raw IC50 Values as Output
2.2. log IC50 Values as Output
2.3. Baseline Performance
3. Discussion
Future Work
4. Methods
4.1. Data
4.1.1. Data Collection
4.1.2. Data Representation
4.2. Network Architectures
4.2.1. Multi-Input Recurrent Neural Network with One-Hot Encoding
4.2.2. Multi-Input Recurrent Neural Network with Embedding
4.2.3. Baseline
4.3. Implementation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASCII | American Standard Code for Information Interchange |
| CNN | Convolutional Neural Network |
| GCNN | Graph Convolutional Neural Network |
| InChI | IUPAC International Chemical Identifier |
| ReLU | Rectified Linear Unit |
| RNN | Recurrent Neural Network |
| SMILES | Simplified Molecular-Input Line-Entry System |
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R & D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed] [Green Version]
- Carpenter, K.A.; Cohen, D.S.; Jarrell, J.T.; Huang, X. Deep learning and virtual drug screening. Future Med. Chem. 2018, 10, 2557–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Korkmaz, S.; Zararsiz, G.; Goksuluk, D. MLViS: A Web Tool for Machine Learning-Based Virtual Screening in Early-Phase of Drug Discovery and Development. PLoS ONE 2015, 10, e0124600. [Google Scholar] [CrossRef]
- Xie, H.; Wen, H.; Zhang, D.; Liu, L.; Liu, B.; Liu, Q.; Jin, Q.; Ke, K.; Hu, M.; Chen, X. Designing of dual inhibitors for GSK-3β and CDK5: Virtual screening and in vitro biological activities study. Oncotarget 2017, 8, 18118–18128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.; Yoon, S. DeepCCI: End-to-end deep learning for chemical-chemical interaction prediction. arXiv 2017, arXiv:1704.0843217, 203–212. [Google Scholar]
- Hamid, M.-N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019, 35, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Jamali, A.A.; Ferdousi, R.; Razzaghi, S.; Li, J.; Safdari, R.; Ebrahimie, E. DrugMiner: Comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov. Today 2016, 21, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive model of linear antimicrobial peptides active against gram-negative bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef]
- Kundu, I.; Paul, G.; Banerjee, R. A machine learning approach towards the prediction of protein–ligand binding affinity based on fundamental molecular properties. RSC Adv. 2018, 8, 12127–12137. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, K.A.; Huang, X. Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: A review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Kriegeskorte, N.; Golan, T. Neural network models and deep learning. Curr. Biol. 2019, 29, R231–R236. [Google Scholar] [CrossRef]
- Pollastri, G.; Przybylski, D.; Rost, B.; Baldi, P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins Struct. Funct. Bioinform. 2002, 47, 228–235. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Esteban, R.; Jiang, X. Differential gene expression in disease: A comparison between high-throughput studies and the literature. BMC Med. Genom. 2017, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Cichonska, A.; Ravikumar, B.; Allaway, R.J.; Park, S.; Wan, F.; Isayev, O.; Li, S.; Mason, M.J.; Lamb, A.; Jeon, M. Crowdsourced mapping of unexplored target space of kinase inhibitors. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of unreliable Q 2! A comparative study of regression metrics for predictivity assessment of QSAR models. J. Chem. Inf. Model. 2016, 56, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Reitz, K.; Cordasco, I.; Prewitt, N. Requests: Http for Humans. KennethReitz [Internet]. 2014. Available online: https://2.python-requests.org/en/master (accessed on 20 July 2020).
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [Green Version]
- Chollet, F. Keras. 2015. Available online: https://github.com/fchollet/keras (accessed on 20 July 2020).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Epochs | Input Rep. | [Q2 for x-val] |
|---|---|---|---|
| 3000 | 500 | One-hot | [−3.8677] |
| 3000 | 500 | Embed | [−0.3743, −0.3382, −0.3323, −0.3917, −0.2402] |
| 10000 | 200 | One-hot | [0.0510, 0.0005, 0, 0.0671] |
| 10000 | 200 | Embed | [0.0391, 0, 0.0364, −0.0689, −0.0512] |
| Model/Input Rep. | Mean Q2 | Mean Q2F3 |
|---|---|---|
| One-hot | 0.0601 | 0.0714 |
| Embed | 0.0537 | 0.0467 |
| AAindex ID | Description |
|---|---|
| KLEP840101 | Net charge |
| KYTJ820101 | Kyte-Doolittle hydrophobicity |
| FASG760101 | Molecular weight |
| FAUJ880103 | Normalized Van der Waals volume |
| GRAR740102 | Polarity |
| CHAM820101 | Polarizability |
| JANJ780101 | Average accessible surface area |
| PRAM900102 | Relative frequency in alpha-helix |
| PRAM900103 | Relative frequency in beta-sheet |
| PRAM900104 | Relative frequency in reverse-turn |
| NADH010104 | Hydropathy scale (20% accessibility) |
| NADH010107 | Hydropathy scale (50% accessibility) |
| RADA880103 | Transfer free energy from vap to chx |
| RICJ880113 | Relative preference value at C2 |
| RICJ880112 | Relative preference value at C3 |
| KHAG800101 | Kerr-constant increments |
| PRAM820103 | Correlation coefficient in regression analysis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpenter, K.; Pilozzi, A.; Huang, X. A Pilot Study of Multi-Input Recurrent Neural Networks for Drug-Kinase Binding Prediction. Molecules 2020, 25, 3372. https://doi.org/10.3390/molecules25153372
Carpenter K, Pilozzi A, Huang X. A Pilot Study of Multi-Input Recurrent Neural Networks for Drug-Kinase Binding Prediction. Molecules. 2020; 25(15):3372. https://doi.org/10.3390/molecules25153372
Chicago/Turabian StyleCarpenter, Kristy, Alexander Pilozzi, and Xudong Huang. 2020. "A Pilot Study of Multi-Input Recurrent Neural Networks for Drug-Kinase Binding Prediction" Molecules 25, no. 15: 3372. https://doi.org/10.3390/molecules25153372
APA StyleCarpenter, K., Pilozzi, A., & Huang, X. (2020). A Pilot Study of Multi-Input Recurrent Neural Networks for Drug-Kinase Binding Prediction. Molecules, 25(15), 3372. https://doi.org/10.3390/molecules25153372