
The Cyclic Nitronate Route to Pharmaceutical Molecules: Synthesis of GSK’s Potent PDE4 Inhibitor as a Case Study


Abstract
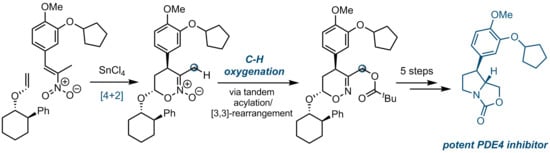
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Denmark, S.E.; Thorarensen, A. Tandem [4 + 2]/[3 + 2] Cycloadditions of Nitroalkenes. Chem. Rev. 1996, 96, 137–166. [Google Scholar] [CrossRef] [PubMed]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L.; Dilman, A.D. Recent Advances in the Synthesis and Chemistry of Nitronates. Synthesis 2017, 49, 3255–3268. [Google Scholar] [CrossRef]
- Mukaijo, Y.; Yokoyama, S.; Nishiwaki, N. Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group. Molecules 2020, 25, 2048. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, L.L.; Burrow, R.A.; Pereira, V.L.P. Diastereoselective synthesis of nitroso acetals from (S,E)-γ-aminated nitroalkenes via multicomponent [4 + 2]/[3 + 2] cycloadditions promoted by LiCl or LiClO4. Beilstein J. Org. Chem. 2013, 9, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Yamamoto, A.; Song, S.; Maruoka, K. Catalytic asymmetric syntheses of isoxazoline-N-oxides under phase-transfer conditions. Chem. Commun. 2011, 47, 4358–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koc, E.; Kwon, O. Total syntheses of heliotridane and pseudoheliotridane through nitrodiene–acrylate 6π-electrocyclization/[3+2] cycloaddition. Tetrahedron 2017, 73, 4195–4200. [Google Scholar] [CrossRef]
- Creech, G.S.; Kwon, O. Tandem 6π-Electrocyclization and Cycloaddition of Nitrodienes to Yield Multicyclic Nitroso Acetals. J. Am. Chem. Soc. 2010, 132, 8876–8877. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-Y.; Deng, X.-M.; Sun, X.-L.; Zheng, J.-C.; Tang, Y. Highly enantioselective synthesis of isoxazoline N-oxides. Chem. Commun. 2008, 6, 738–740. [Google Scholar] [CrossRef]
- Streitferdt, V.; Haindl, M.H.; Hioe, J.; Morana, F.; Renzi, P.; von Rekowski, F.; Zimmermann, A.; Nardi, M.; Zeitler, K.; Gschwind, R.M. Unprecedented Mechanism of an Organocatalytic Route to Conjugated Enynes with a Junction to Cyclic Nitronates. Eur. J. Org. Chem. 2019, 2019, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Elsner, P.; Jensen, K.L.; Falcicchio, A.; Marcos, V.; Jørgensen, K.A. Achieving Molecular Complexity by Organocatalytic One-Pot Strategies—A Fast Entry for Synthesis of Sphingoids, Amino Sugars, and Polyhydroxylated α-Amino Acids. Angew. Chem. Int. Ed. 2009, 48, 6844–6848. [Google Scholar] [CrossRef]
- Baiazitov, R.Y.; Denmark, S.E. Tandem [4 + 2]/[3 + 2] Cycloadditions. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 471–550. [Google Scholar]
- Denmark, S.E.; Martinborough, E.A. Enantioselective Total Syntheses of (+)-Castanospermine, (+)-6-Epicastanospermine, (+)-Australine, and (+)-3-Epiaustraline. J. Am. Chem. Soc. 1999, 121, 3046–3056. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A.; Middleton, D.S. Tandem [4 + 2]/[3 + 2] Cycloadditions of Nitroalkenes. 9. Synthesis of (−)-Rosmarinecine. J. Am. Chem. Soc. 1996, 118, 8266–8277. [Google Scholar] [CrossRef]
- Denmark, S.E.; Baiazitov, R.Y.; Nguyen, S.T. Tandem double intramolecular [4 + 2]/[3 + 2] cycloadditions of nitroalkenes: Construction of the pentacyclic core structure of daphnilactone B. Tetrahedron 2009, 65, 6535–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmark, S.E.; Montgomery, J.I.; Kramps, L.A. Synthesis, X-ray Crystallography, and Computational Analysis of 1-Azafenestranes. J. Am. Chem. Soc. 2006, 128, 11620–11630. [Google Scholar] [CrossRef]
- Denmark, S.E.; Montgomery, J.I. Synthesis of cis,cis,cis,cis-[5.5.5.4]-1-Azafenestrane with Discovery of an Unexpected Dyotropic Rearrangement. Angew. Chem. Int. Ed. 2005, 44, 3732–3736. [Google Scholar] [CrossRef]
- For a review see: Sukhorukov, A.Y. C-H Reactivity of the α-Position in Nitrones and Nitronates. Adv. Synth. Catal. 2020, 362, 724–754. [Google Scholar] [CrossRef]
- Tishkov, A.A.; Lesiv, A.V.; Khomutova, Y.A.; Strelenko, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Ioffe, S.L.; Denmark, S.E. 2-Silyloxy-1,2-oxazines, a New Type of Acetals of Conjugated Nitroso Alkenes. J. Org. Chem. 2003, 68, 9477–9480. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y.; Kapatsyna, M.A.; Yi, T.L.T.; Park, H.R.; Naumovich, Y.A.; Zhmurov, P.A.; Khomutova, Y.A.; Ioffe, S.L.; Tartakovsky, V.A. A General Metal-Assisted Synthesis of α-Halo Oxime Ethers from Nitronates and Nitro Compounds. Eur. J. Org. Chem. 2014, 2014, 8148–8159. [Google Scholar] [CrossRef]
- Naumovich, Y.A.; Buckland, V.E.; Sen’ko, D.A.; Nelyubina, Y.V.; Khoroshutina, Y.A.; Sukhorukov, A.Y.; Ioffe, S.L. Metal-assisted addition of a nitrate anion to bis(oxy)enamines. A general approach to the synthesis of α-nitroxy-oxime derivatives from nitronates. Org. Biomol. Chem. 2016, 14, 3963–3974. [Google Scholar] [CrossRef] [Green Version]
- Tabolin, A.A.; Lesiv, A.V.; Khomutova, Y.A.; Nelyubina, Y.V.; Ioffe, S.L. Rearrangement of 3-alkylidene-2-siloxy-tetrahydro-1,2-oxazines (ASENA). A new approach toward the synthesis of 3-α-hydroxyalkyl-5,6-dihydro-4H-1,2-oxazines. Tetrahedron 2009, 65, 4578–4592. [Google Scholar] [CrossRef]
- Naumovich, Y.A.; Golovanov, I.S.; Sukhorukov, A.Y.; Ioffe, S.L. Addition of HO-Acids to N,N-Bis(oxy)enamines: Mechanism, Scope and Application to the Synthesis of Pharmaceuticals. Eur. J. Org. Chem. 2017, 2017, 6209–6227. [Google Scholar] [CrossRef]
- Zhmurov, P.A.; Khoroshutina, Y.A.; Novikov, R.A.; Golovanov, I.S.; Sukhorukov, A.Y.; Ioffe, S.L. Divergent Reactivity of In Situ Generated Metal Azides: Reaction with N,N-Bis(oxy)enamines as a Case Study. Chem. Eur. J. 2017, 23, 4570–4578. [Google Scholar] [CrossRef] [PubMed]
- Zhmurov, P.A.; Sukhorukov, A.Y.; Chupakhin, V.I.; Khomutova, Y.V.; Ioffe, S.L.; Tartakovsky, V.A. Synthesis of PDE IV inhibitors. First asymmetric synthesis of two of GlaxoSmithKline’s highly potent Rolipram analogues. Org. Biomol. Chem. 2013, 11, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, A.Y.; Boyko, Y.D.; Ioffe, S.L.; Khomutova, Y.A.; Nelyubina, Y.V.; Tartakovsky, V.A. Synthesis of PDE IVb Inhibitors. 1. Asymmetric Synthesis and Stereochemical Assignment of (+)- and (−)-7-[3-(Cyclopentyloxy)-4-methoxyphenyl]hexahydro-3H-pyrrolizin-3-one. J. Org. Chem. 2011, 76, 7893–7900. [Google Scholar] [CrossRef] [PubMed]
- Kokuev, A.O.; Antonova, Y.A.; Dorokhov, V.S.; Golovanov, I.S.; Nelyubina, Y.V.; Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L. Acylation of Nitronates: [3,3]-Sigmatropic Rearrangement of in Situ Generated N-Acyloxy,N-oxyenamines. J. Org. Chem. 2018, 83, 11057–11066. [Google Scholar] [CrossRef]
- Brackeen, M.F.; Stafford, J.A.; Cowan, D.J.; Brown, P.J.; Domanico, P.L.; Feldman, P.L.; Rose, D.; Strickland, A.B.; Veal, J.M.; Verghese, M. Design and Synthesis of Conformationally Constrained Analogs of 4-(3-Butoxy-4-methoxybenzyl)imidazolidin-2-one (Ro 20-1724) as Potent Inhibitors of cAMP-Specific Phosphodiesterase. J. Med. Chem. 1995, 38, 4848–4854. [Google Scholar] [CrossRef]
- Jackson, E.K.; Carcillo, J.A. Treatment of Sepsis-Induced Acute Renal Failure. U.S. Patent 5849774, 15 December 1998. [Google Scholar]
- Mulhall, A.M.; Droege, C.A.; Ernst, N.E.; Panos, R.J.; Zafar, M.A. Phosphodiesterase 4 inhibitors for the treatment of chronic obstructive pulmonary disease: A review of current and developing drugs. Exp. Opin. Investig. Drugs 2015, 24, 1597–1611. [Google Scholar] [CrossRef]
- Zhmurov, P.A.; Tabolin, A.A.; Sukhorukov, A.Y.; Lesiv, A.V.; Klenov, M.S.; Khomutova, Y.A.; Ioffe, S.L.; Tartakovsky, V.A. Synthesis of phosphodiesterase IVb inhibitors 2. Stereoselective synthesis of hexahydro-3H-pyrrolo[1,2-c]imidazol-3-one and tetrahydro-1H-pyrrolo[1,2-c][1,3]oxazol-3-one derivatives. Russ. Chem. Bull. 2011, 60, 2390–2395. [Google Scholar] [CrossRef]
- Yasuo, T.; Norihiko, Y.; Hiroko, M.; Toshihide, K.; Masahiro, A.; Yutaka, M. Synthesis and a Novel Fragmentation of 6-Alkoxy-5,6-dihydro-4H-1,2-oxazine 2-Oxide. Bull. Chem. Soc. Jpn. 1988, 61, 461–465. [Google Scholar]
- Sukhorukov, A.Y.; Ioffe, S.L. Chemistry of Six-Membered Cyclic Oxime Ethers. Application in the Synthesis of Bioactive Compounds. Chem. Rev. 2011, 111, 5004–5041. [Google Scholar] [CrossRef]
- Zimmer, R.; Arnold, T.; Homann, K.; Reissig, H.-U. An Efficient and Simple Synthesis of 3,4,5,6-Tetrahydro-2H-1,2-oxazines by Sodium Cyanoborohydride Reduction of 5,6-Dihydro-4H-1,2-oxazines. Synthesis 1994, 1994, 1050–1056. [Google Scholar] [CrossRef]
- Demchenko, A.V.; Rousson, E.; Boons, G.-J. Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar] [CrossRef]
- Ma, Y.; Lian, G.; Li, Y.; Yu, B. Identification of 3,6-di-O-acetyl-1,2,4-O-orthoacetyl-α-d-glucopyranose as a direct evidence for the 4-O-acyl group participation in glycosylation. Chem. Commun. 2011, 47, 7515–7517. [Google Scholar] [CrossRef] [PubMed]
- Beaver, M.G.; Billings, S.B.; Woerpel, K.A. C-Glycosylation Reactions of Sulfur-Substituted Glycosyl Donors: Evidence against the Role of Neighboring-Group Participation. J. Am. Chem. Soc. 2008, 130, 2082–2086. [Google Scholar] [CrossRef] [PubMed]
- Stalford, S.A.; Kilner, C.A.; Leach, A.G.; Turnbull, W.B. Neighbouring group participation vs. addition to oxacarbenium ions: Studies on the synthesis of mycobacterial oligosaccharides. Org. Biomol. Chem. 2009, 7, 4842–4852. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yang, W.; Ramadan, S.; Huang, X. Pre-Activation-Based Stereoselective Glycosylations. Eur. J. Org. Chem. 2018, 2018, 1075–1096. [Google Scholar] [CrossRef]
- Crich, D.; Hu, T.; Cai, F. Does Neighboring Group Participation by Non-Vicinal Esters Play a Role in Glycosylation Reactions? Effective Probes for the Detection of Bridging Intermediates. J. Org. Chem. 2008, 73, 8942–8953. [Google Scholar] [CrossRef] [Green Version]
- Komarova, B.S.; Tsvetkov, Y.E.; Nifantiev, N.E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. [Google Scholar] [CrossRef]
Sample Availability: Samples of racemic and enantiopure PDE4 inhibitor CMPO are available from the authors. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospelov, E.V.; Golovanov, I.S.; Ioffe, S.L.; Sukhorukov, A.Y. The Cyclic Nitronate Route to Pharmaceutical Molecules: Synthesis of GSK’s Potent PDE4 Inhibitor as a Case Study. Molecules 2020, 25, 3613. https://doi.org/10.3390/molecules25163613
Pospelov EV, Golovanov IS, Ioffe SL, Sukhorukov AY. The Cyclic Nitronate Route to Pharmaceutical Molecules: Synthesis of GSK’s Potent PDE4 Inhibitor as a Case Study. Molecules. 2020; 25(16):3613. https://doi.org/10.3390/molecules25163613
Chicago/Turabian StylePospelov, Evgeny V., Ivan S. Golovanov, Sema L. Ioffe, and Alexey Yu. Sukhorukov. 2020. "The Cyclic Nitronate Route to Pharmaceutical Molecules: Synthesis of GSK’s Potent PDE4 Inhibitor as a Case Study" Molecules 25, no. 16: 3613. https://doi.org/10.3390/molecules25163613
APA StylePospelov, E. V., Golovanov, I. S., Ioffe, S. L., & Sukhorukov, A. Y. (2020). The Cyclic Nitronate Route to Pharmaceutical Molecules: Synthesis of GSK’s Potent PDE4 Inhibitor as a Case Study. Molecules, 25(16), 3613. https://doi.org/10.3390/molecules25163613