
In Silico Identification of Potential Natural Product Inhibitors of Human Proteases Key to SARS-CoV-2 Infection

 ,
,  , ,
, ,  and
and
Abstract
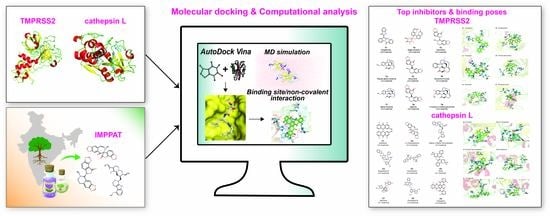
:
1. Introduction
2. Results
2.1. Workflow for Virtual Screening
2.2. Potential Phytochemical Inhibitors of TMPRSS2
2.3. Potential Phytochemical Inhibitors of Cathepsin L
2.4. Molecular Dynamics Simulation of Top Inhibitors
2.5. MM-PBSA Binding Energy of Top Inhibitors
3. Materials and Methods
3.1. Phytochemical Library and Drug-Likeness Evaluation
3.2. Homology Modeling of TMPRSS2 Structure
3.3. Protein Structure of Cathepsin L
3.4. Molecular Docking
3.5. Identification of Protein-Ligand Interactions
3.6. Comparison with Reference Inhibitors of TMPRSS2 and Cathepsin L
3.7. Molecular Dynamics Simulations
3.8. MM-PBSA Calculation
3.9. Validation of the TMPRSS2 Model Structure
3.10. Prediction of ADMET Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamic |
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| TMPRSS2 | Transmembrane Protease Serine 2 |
| RBD | Receptor binding domain |
| ACE-2 | Angiotensin converting enzyme 2 |
| ORF | Open reading frame |
| FDA | Food and drug administration |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| MM-PBSA | Molecular Mechanics Poisson-Boltzmann Surface Area |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Li, W. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Genet. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, 64. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- ElFiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef]
- Islam, R.; Parves, R.; Paul, A.S.; Uddin, N.; Rahman, S.; Al Mamun, A.; Hossain, N.; Ali, A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Q.; Wang, Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics 2020, 36, 3295–3298. [Google Scholar] [CrossRef] [PubMed]
- Olubiyi, O.O.; Olagunju, M.O.; Keutmann, M.; Loschwitz, J.; Strodel, B. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Molecules 2020, 25, 3193. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Dwivedy, A.; Mariadasse, R.; Tiwari, S.; Kar, D.; Jeyakanthan, J.; Biswal, B.K. Prediction of Small Molecule Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus-2 RNA-dependent RNA Polymerase. ACS Omega 2020. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.-L.; Zhang, A.-H.; Wang, X.-J. Traditional Chinese medicine for COVID-19 treatment. Pharmacol. Res. 2020, 155, 104743. [Google Scholar] [CrossRef]
- Vellingiri, B.; Jayaramayya, K.; Iyer, M.; Narayanasamy, A.; Govindasamy, V.; Giridharan, B.; Ganesan, S.; Venugopal, A.; Venkatesan, D.; Ganesan, H.; et al. COVID-19: A promising cure for the global panic. Sci. Total Environ. 2020, 725, 138277. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 Gene, Which Encodes a Novel Serine Protease with Transmembrane, LDLRA, and SRCR Domains and Maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Lee, C.-L.; Yen, H.-R.; Chang, Y.-S.; Lin, Y.-P.; Huang, S.-H.; Lin, C.-W. Antiviral Action of Tryptanthrin Isolated from Strobilanthes cusia Leaf against Human Coronavirus NL63. Biomolecules 2020, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.-G.; Zhou, A.-Y.; Du, Z.; Zhang, Y.; Zhang, K.; Ma, Y.-Y. Coumarins with α-glucosidase and α-amylase inhibitory activities from the flower of Edgeworthia gardneri. Fitoterapia 2015, 107, 122–127. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.-L.; Xu, P.; Lin, Y.-X.; Yang, F.; Liu, J.-H.; Zhu, H.-W.; Xia, Z. In vitro evaluation of dual agonists for PPARγ/β from the flower of Edgeworthia gardneri (wall.) Meisn. J. Ethnopharmacol. 2015, 162, 14–19. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.-L.; Yang, F.; Li, F.; Zhang, Q.-H.; Xia, Z.-N. The flower of Edgeworthia gardneri (wall.) Meisn. suppresses adipogenesis through modulation of the AMPK pathway in 3T3-L1 adipocytes. J. Ethnopharmacol. 2016, 191, 379–386. [Google Scholar] [CrossRef]
- Gupta, P.C.; Sharma, N.; Rao, C.V. A review on ethnobotany, phytochemistry and pharmacology of Fumaria indica (Fumitory). Asian Pac. J. Trop. Biomed. 2012, 2, 665–669. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Blaskó, G.; Kerekes, P.; Kovács, I.; Simonyi, M. Inhibition of [3H]GABA binding to rat brain synaptic membranes by bicuculline related alkaloids. Biochem. Pharmacol. 1984, 33, 3537–3545. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials. Acc. Chem. Res. 2020. [Google Scholar] [CrossRef]
- Acharya, R.; Mitra, S.; Shukla, V. Effect of Shodhana (processing) on Kupeelu (Strychnos nux-vomica Linn.) with special reference to strychnine and brucine content. AYU (An. Int. Q. J. Res. Ayurveda) 2011, 32, 402–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, S.; Bracamontes, J.; Zorumski, C.; Weiss, D.S.; Steinbach, J.H. Bicuculline and Gabazine Are Allosteric Inhibitors of Channel Opening of the GABAA Receptor. J. Neurosci. 1997, 17, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, C.P. Indian Medicinal Plants: An Illustrated Dictionary; Springer: New York, NY, USA, 2007. [Google Scholar]
- Yadav, J.P.; Arya, V.; Yadav, S.; Panghal, M.; Kumar, S.; Dhankhar, S. Cassia occidentalis L.: A review on its ethnobotany, phytochemical and pharmacological profile. Fitoterapia 2010, 81, 223–230. [Google Scholar] [CrossRef]
- Zhu, S.H.; Yang, Z.Q.; Peng, X.L. Application of Anthraqinone Derivative in Resisting Influenza Virus and Bird Flu Virus H5N1. China Patent CN20051134677, 21 December 2005. [Google Scholar]
- Mete, I.E.; Gözler, T. (+)-Oxoturkiyenine: An Isoquinoline-Derived Alkaloid from Hypecoum pendulum. J. Nat. Prod. 1988, 51, 272–274. [Google Scholar] [CrossRef]
- Gurung, P.; De, P. Spectrum of biological properties of cinchona alkaloids: A brief review. J. Pharmacogn. Phytochem. 2017, 6, 162–166. [Google Scholar]
- Tripathi, Y.C.; Maurya, S.; Singh, V.; Pandey, V. Cyclopeptide alkaloids from Zizyphus rugosa bark. Phytochemistry 1989, 28, 1563–1565. [Google Scholar] [CrossRef]
- Yadav, A.; Singh, P. Analgesic and anti-inflammatory activities of Zizyphus rugosa root barks. J. Chem. Pharm. Res. 2010, 2, 255–259. [Google Scholar]
- Wang, J.-H.; Luan, F.; He, X.-D.; Wang, Y.; Li, M.-X. Traditional uses and pharmacological properties of Clerodendrum phytochemicals. J. Tradit. Complement. Med. 2018, 8, 24–38. [Google Scholar] [CrossRef]
- Sumthong, P.; Romero-González, R.R.; Verpoorte, R. Identification of Anti-Wood Rot Compounds in Teak (Tectona grandisL.f.) Sawdust Extract. J. Wood Chem. Technol. 2008, 28, 247–260. [Google Scholar] [CrossRef]
- Jain, M.; Kapadia, R.; Jadeja, R.N.; Thounaojam, M.C.; Devkar, R.V.; Mishra, S.H. Traditional uses, phytochemistry and pharmacology of Tecomella undulata—A review. Asian Pac. J. Trop. Biomed. 2012, 2, 1918. [Google Scholar] [CrossRef]
- Cadelis, M.; Bourguet-Kondracki, M.-L.; Dubois, J.; Valentin, A.; Barker, D.; Copp, B.R. Discovery and preliminary structure–activity relationship studies on tecomaquinone I and tectol as novel farnesyltransferase and plasmodial inhibitors. Bioorganic Med. Chem. 2016, 24, 3102–3107. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, E. Effect of the flavanolignans of Silybum marianum L. On lipid peroxidation in rat liver microsomes and freshly isolated hepatocytes. Pharmacol. Res. 1992, 25, 147–165. [Google Scholar] [CrossRef]
- Bahmani, M.; Shirzad, H.; Rafieian, S.; Rafieian-Kopaei, M. Silybum marianum: Beyond Hepatoprotection. J. Evid. -Based Complementary Altern. Med. 2015, 20, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. Open Source Drug Discovery Consortium g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- The Wealth of India the wealth of India: A Dictionary of Indian Raw Materials & Industrial Products. First Supplement Series (Raw Materials); Publications & Information Directorate, Council of Scientific & Industrial Research: New Delhi, India, 2000; Volume 1.
- The Wealth of India the Wealth of India: A Dictionary of Indian Raw Materials & Industrial Products. First Supplement Series (Raw Materials); Publications & Information Directorate, Council of Scientific & Industrial Research: New Delhi, India, 2000; Volume 2.
- The Wealth of India the wealth of India: A Dictionary of Indian Raw Materials & Industrial Products. First Supplement Series (Raw Materials); Publications & Information Directorate, Council of Scientific & Industrial Research: New Delhi, India, 2000; Volume 3.
- The Wealth of India the Wealth of India: A Dictionary of Indian Raw Materials & Industrial Products. First Supplement Series (Raw Materials); Publications & Information Directorate, Council of Scientific & Industrial Research: New Delhi, India, 2000; Volume 4.
- The Wealth of India the Wealth of India: A Dictionary of Indian Raw Materials & Industrial Products. First Supplement Series (Raw Materials); Publications & Information Directorate, Council of Scientific & Industrial Research: New Delhi, India, 2000; Volume 5.
- Rastogi, R.P. (Ed.) Drug Research Perspectives. In Compendium of Indian Medicinal Plants; Central Drug Research Institute: Lucknow, India, 1990; Volume 1, pp. 1960–1969. [Google Scholar]
- Rastogi, R.P. (Ed.) Drug Research Perspectives. In Compendium of Indian Medicinal Plants; Central Drug Research Institute: Lucknow, India, 1991; Volume 2, pp. 1970–1979. [Google Scholar]
- Rastogi, R.P. (Ed.) Drug Research Perspectives. In Compendium of Indian Medicinal Plants; Central Drug Research Institute: Lucknow, India, 1993; Volume 3, pp. 1980–1984. [Google Scholar]
- Rastogi, R.P. (Ed.) Drug Research Perspectives. In Compendium of Indian Medicinal Plants; Central Drug Research Institute: Lucknow, India, 1995; Volume 4, pp. 1985–1989. [Google Scholar]
- Rastogi, R.P. (Ed.) Drug Research Perspectives. In Compendium of Indian Medicinal Plants; Central Drug Research Institute: Lucknow, India, 1998; Volume 5, pp. 1990–1994. [Google Scholar]
- Evnin, L.B.; Vasquez, J.R.; Craik, C.S. Substrate specificity of trypsin investigated by using a genetic selection. Proc. Natl. Acad. Sci. USA 1990, 87, 6659–6663. [Google Scholar] [CrossRef] [Green Version]
- Herter, S.; Piper, D.E.; Aaron, W.; Gabriele, T.; Cutler, G.; Cao, P.; Bhatt, A.S.; Choe, Y.; Craik, C.S.; Walker, N.; et al. Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers. Biochem. J. 2005, 390, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, B.; Tichý, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Imai, Y.; Nomura, T.; Fujisawa, Y.; Yamamoto, Y.; Sugawara, T. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 1997, 407, 47–50. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams-Cioaba, M.A.; Krupa, J.C.; Xu, C.; Mort, J.S.; Min, J. Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat. Commun. 2011, 2, 197. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.P.; Mc Teixeira, J.; Trellet, M.; Bonvin, A.M.J.J. pdb-tools: A swiss army knife for molecular structures. F1000Research 2018, 7, 1961. [Google Scholar] [CrossRef]
- Schmidt, T.; Haas, J.; Cassarino, T.G.; Schwede, T. Assessment of ligand-binding residue predictions in CASP9. Proteins Struct. Funct. Bioinform. 2011, 79, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Sarkhel, S.; Desiraju, G.R. N-H…O, O-H…O, and C-H…O hydrogen bonds in protein-ligand complexes: Strong and weak interactions in molecular recognition. Proteins Struct. Funct. Bioinform. 2003, 54, 247–259. [Google Scholar] [CrossRef]
- Zhou, P.; Tian, F.; Lv, F.; Shang, Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins Struct. Funct. Bioinform. 2008, 76, 151–163. [Google Scholar] [CrossRef]
- Kříž, K.; Fanfrlik, J.; Lepsik, M. Chalcogen Bonding in Protein−Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. Chem. Phys. Chem. 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Borozan, S.; Stojanović, S.Đ. Halogen bonding in complexes of proteins and non-natural amino acids. Comput. Biol. Chem. 2013, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB† †Electronic supplementary information (ESI) available. See doi:10.1039/c7md00381a. Med. Chem. Comm. 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Turk, V.; Turk, B.; Turk, D. NEW EMBO MEMBERS’ REVIEW: Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 2001, 20, 4629–4633. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Luo, S.; Libby, P.; Shi, G. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytochemical Symbol | Binding Energy (kcal/mol) | Common Name | Plant Source |
|---|---|---|---|
| T1 | −9.6 | Qingdainone | Strobilanthes cusia[*] |
| T2 | −9.6 | Edgeworoside C | Edgeworthia gardneri |
| T3 | −9.6 | Adlumidine | Fumaria indica[*] |
| T4 | −9.3 | Pseudo-α-Colubrine | Strychnos nux-vomica[*] |
| T5 | −9.3 | Bicuculline | Fumaria indica[*], Corydalis govaniana [*], Nerium oleander [*] |
| T6 | −9.3 | Strychnine N-oxide | Strychnos nux-vomica[*], Strychnos ignatii [*], Strychnos colubrina [*] |
| T7 | −9.2 | α-Colubrine | Strychnos nux-vomica[*], Strychnos ignatii [*], Strychnos colubrina [*] |
| T8 | −9.2 | Egenine | Fumaria vaillantii[*] |
| T9 | −9.2 | 2-Hydroxy-3-methoxystrychnine | Strychnos nux-vomica[*] |
| Phytochemical Symbol | Binding Energy (kcal/mol) | Common Name | Plant Source |
|---|---|---|---|
| C1 | −8.9 | Ararobinol | Senna occidentalis[*] |
| C2 | −8.3 | (+)-Oxoturkiyenine | Hypecoum pendulum |
| C3 | −8.3 | 3Alpha,17Alpha-Cinchophylline | Cinchona calisaya[*] |
| C4 | −8.2 | Rugosanine B | Ziziphus rugosa[*] |
| C5 | −8.2 | Trichotomine | Clerodendrum trichotomum[*] |
| C6 | −8.1 | Tectol | Tectona grandis[*], Tecomella undulata [*] |
| C7 | −8.1 | Silymonin | Silybum marianum[*] |
| C8 | −8 | Picrasidine M | Picrasma quassioides[*] |
| C9 | −8 | Trisjuglone | Juglans regia[*] |
| Protein-Ligand Complex | Binding Energy (kcal/mol) | Van Der Waals Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | Polar Solvation Energy (kcal/mol) | SASA Energy (kcal/mol) |
|---|---|---|---|---|---|
| TMPRSS2-T1 | −39.15 ± 2.799 | −54.285 ± 2.903 | −3.031 ±1.439 | 22.844 ± 2.44 | −4.678 ± 0.237 |
| TMPRSS2-T2 | −30.284 ±3.585 | −49.048 ± 3.838 | −12.501 ± 4.884 | 35.978 ± 5.226 | −4.712 ± 0.320 |
| TMPRSS2-T3 | −27.386 ± 2.077 | −39.379 ± 2.109 | −8.846 ± 1.423 | 24.359 ± 2.157 | −3.52 ± 0.210 |
| cathepsin L-C1 | −22.384 ± 3.420 | −25.296 ± 3.127 | −2.214 ± 1.661 | 7.988 ± 4.103 | −2.861 ± 0.366 |
| cathepsin L-C2 | −20.577 ± 3.600 | −30.129 ± 3.154 | −4.572 ± 2.138 | 16.891 ± 3.533 | −2.767 ± 0.234 |
| cathepsin L-C3 | −26.156 ± 3.433 | −37.165 ± 3.308 | −2.093 ± 1.379 | 16.958 ± 4.513 | −3.856 ± 0.319 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivek-Ananth, R.P.; Rana, A.; Rajan, N.; Biswal, H.S.; Samal, A. In Silico Identification of Potential Natural Product Inhibitors of Human Proteases Key to SARS-CoV-2 Infection. Molecules 2020, 25, 3822. https://doi.org/10.3390/molecules25173822
Vivek-Ananth RP, Rana A, Rajan N, Biswal HS, Samal A. In Silico Identification of Potential Natural Product Inhibitors of Human Proteases Key to SARS-CoV-2 Infection. Molecules. 2020; 25(17):3822. https://doi.org/10.3390/molecules25173822
Chicago/Turabian StyleVivek-Ananth, R.P., Abhijit Rana, Nithin Rajan, Himansu S. Biswal, and Areejit Samal. 2020. "In Silico Identification of Potential Natural Product Inhibitors of Human Proteases Key to SARS-CoV-2 Infection" Molecules 25, no. 17: 3822. https://doi.org/10.3390/molecules25173822
APA StyleVivek-Ananth, R. P., Rana, A., Rajan, N., Biswal, H. S., & Samal, A. (2020). In Silico Identification of Potential Natural Product Inhibitors of Human Proteases Key to SARS-CoV-2 Infection. Molecules, 25(17), 3822. https://doi.org/10.3390/molecules25173822