
Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease


Abstract
:1. Introduction
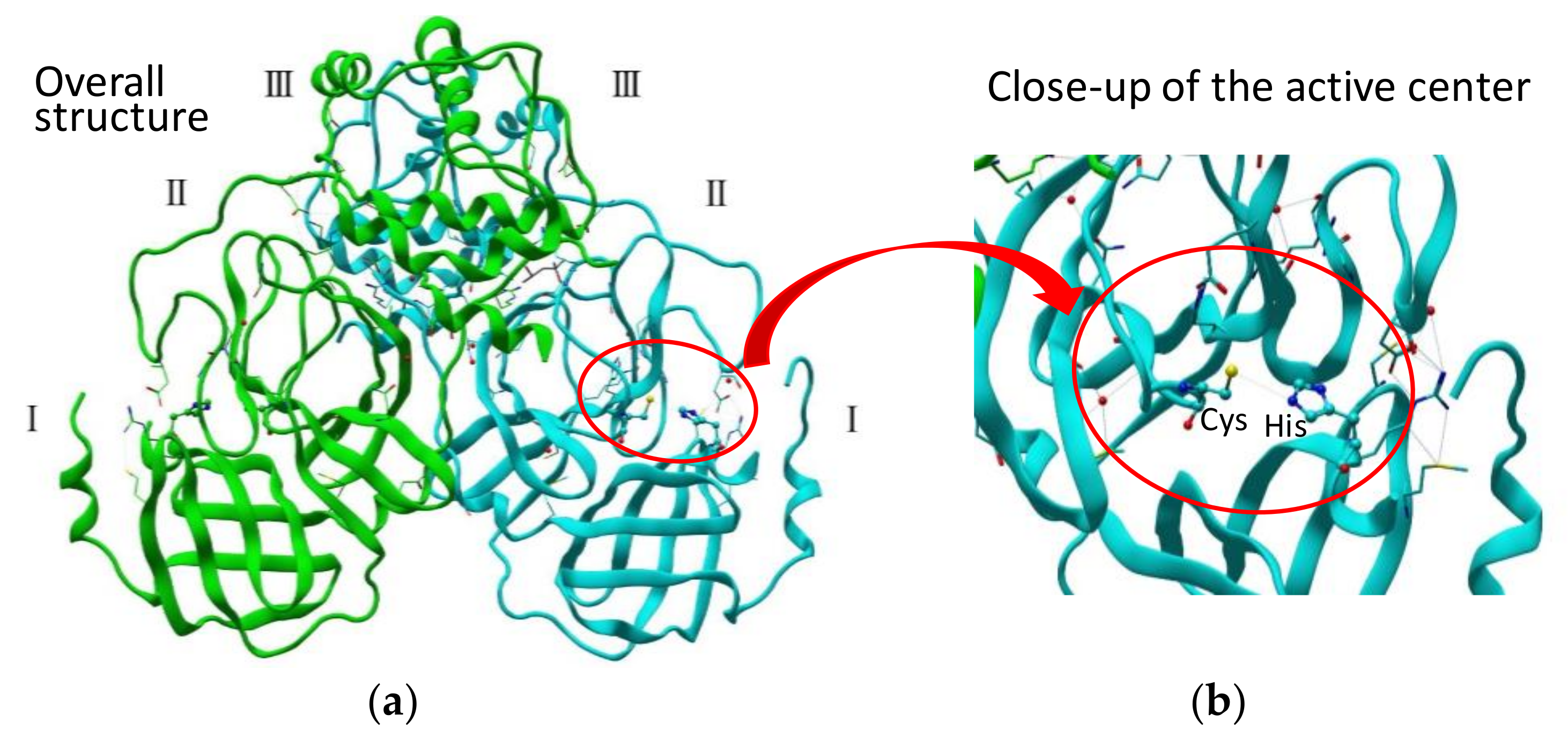
2. SARS 3CL Protease
3. Inhibitors of SARS-CoV 3CLpro
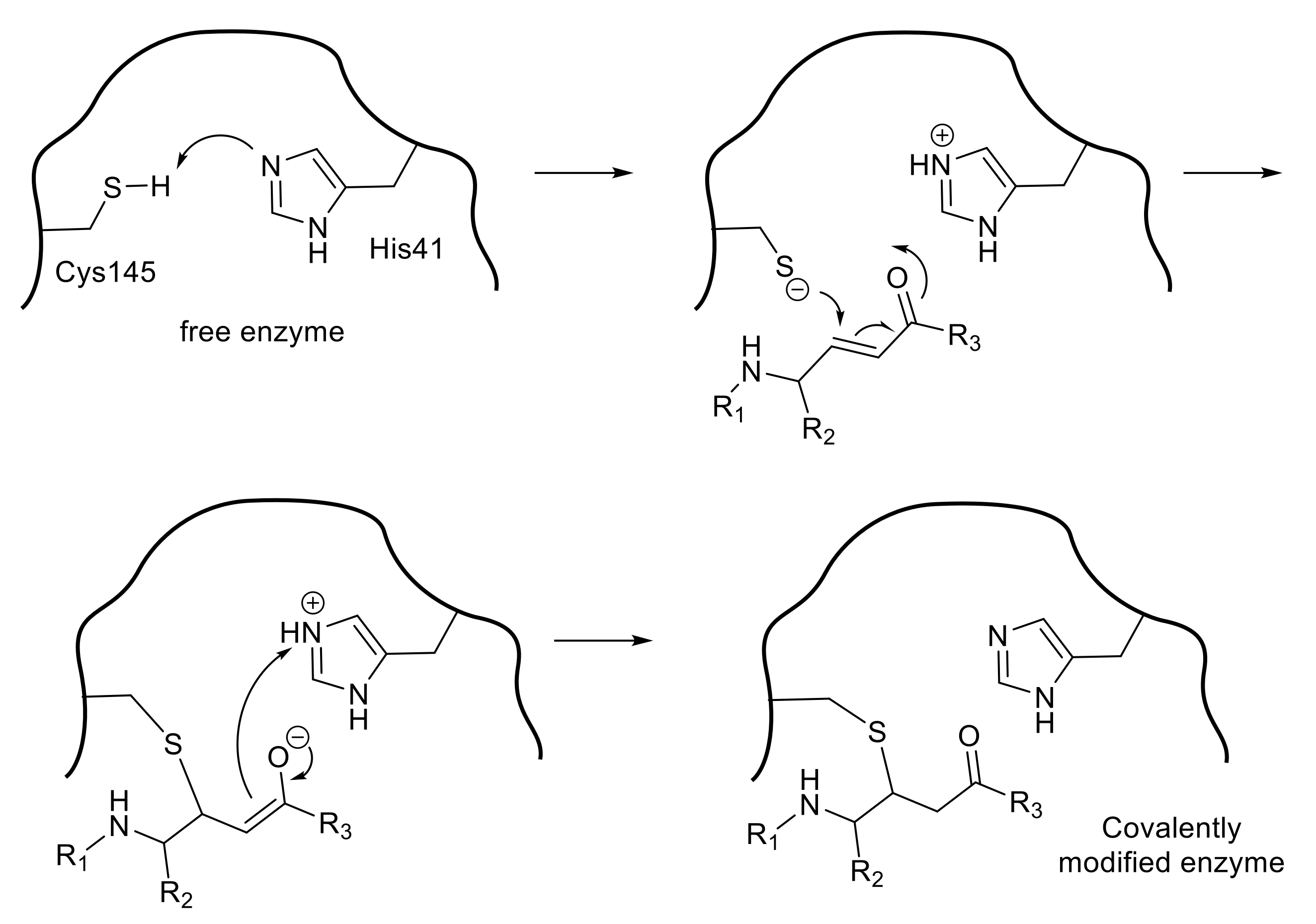
3.1. Peptide-Mimetic Inhibitors Containing a Michael Acceptor
3.2. Peptides with Halomethyl Ketone
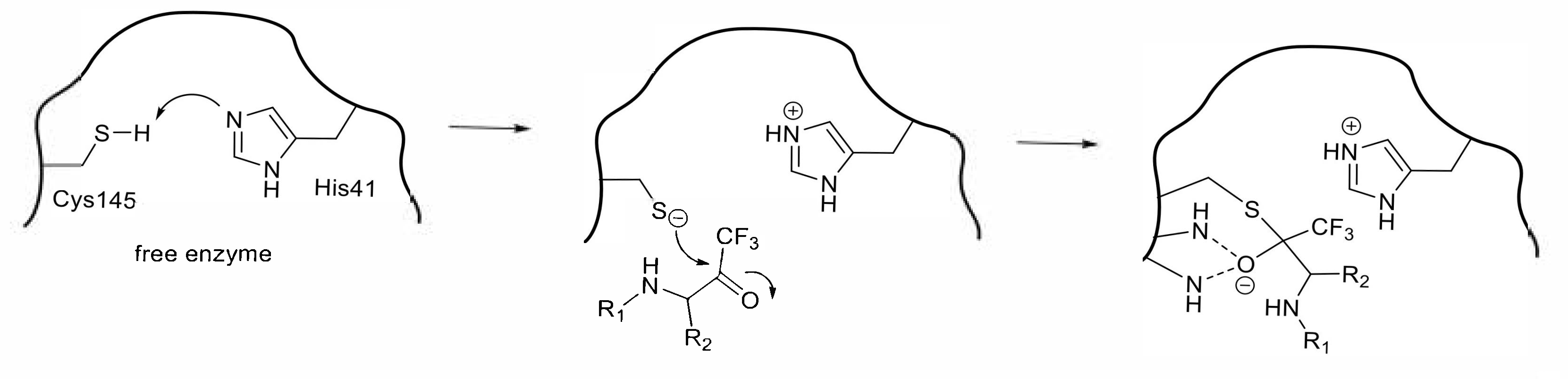
3.3. Peptides with Trifluoromethyl Ketone and Related Electrophilic Substituent
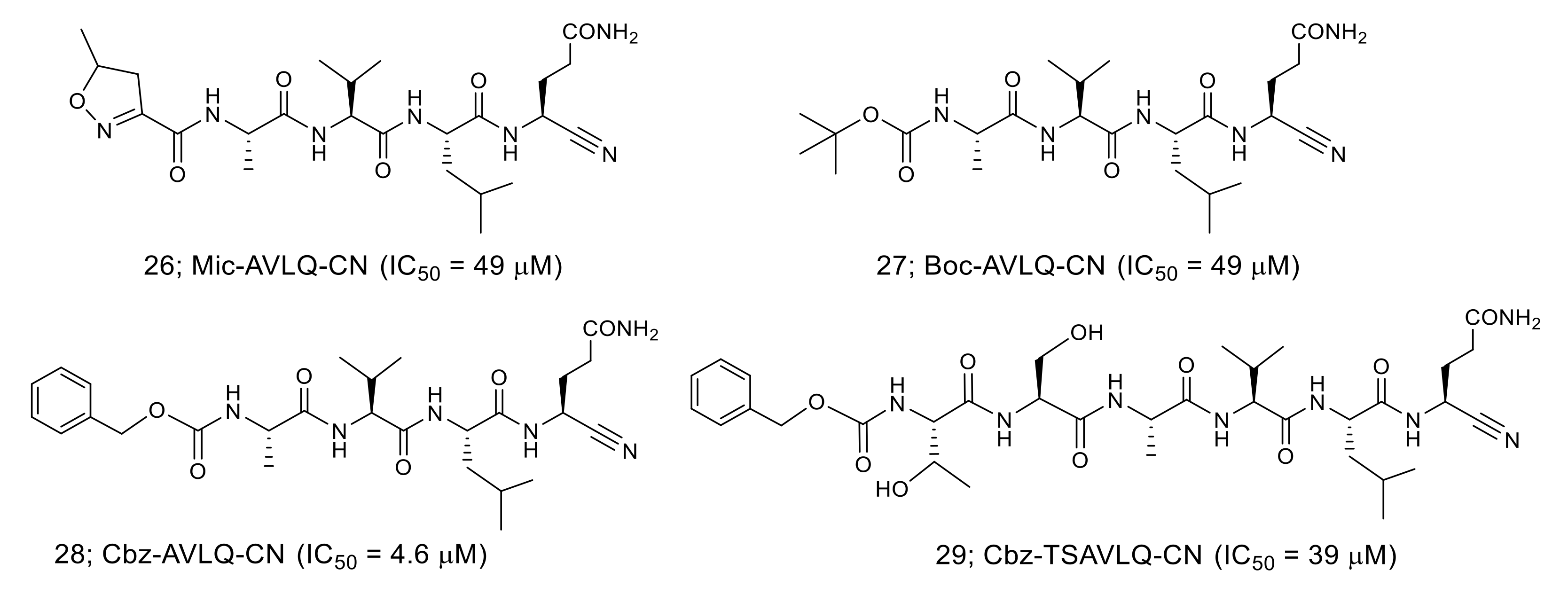
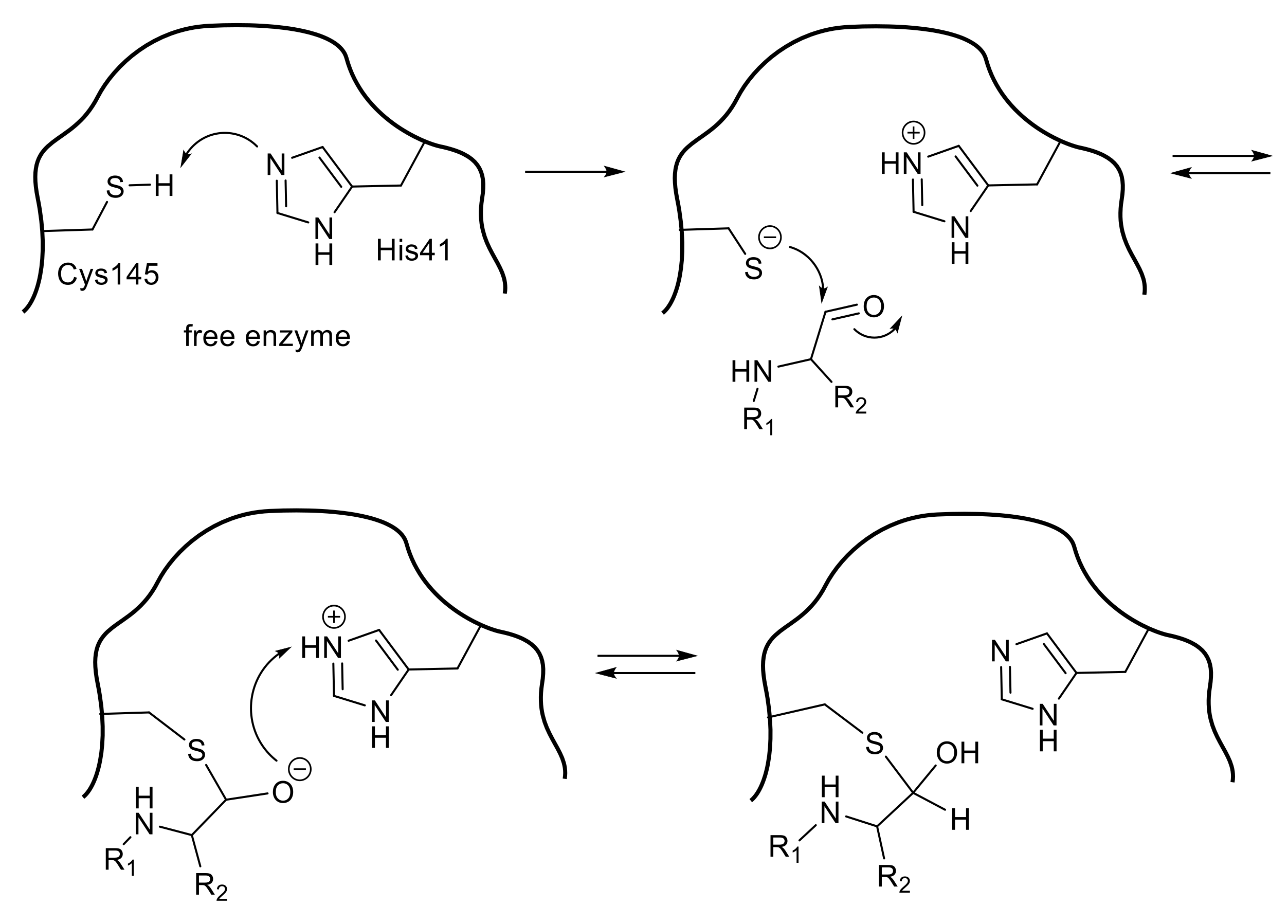
3.4. Nitrile-Based Peptide-Mimetic Inhibitors
3.5. Peptide Aldehydes
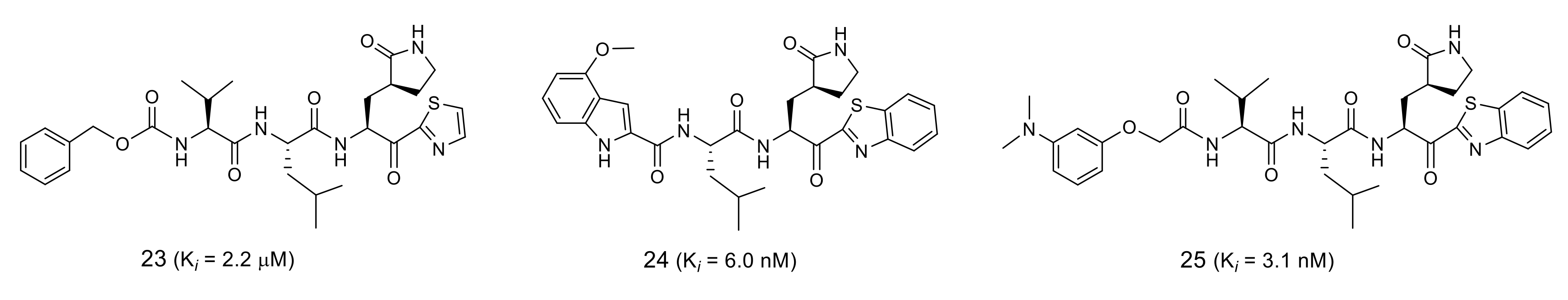
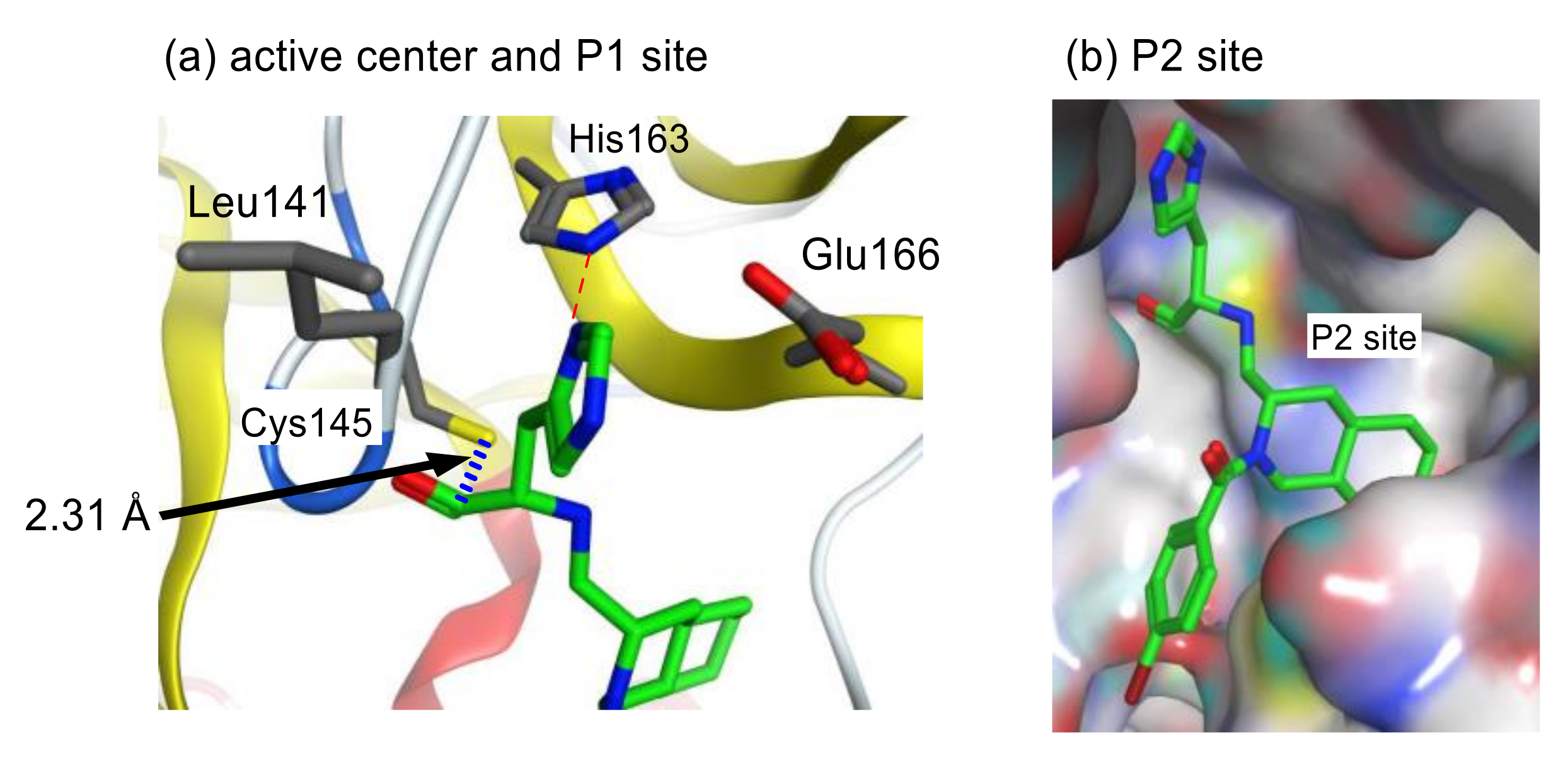
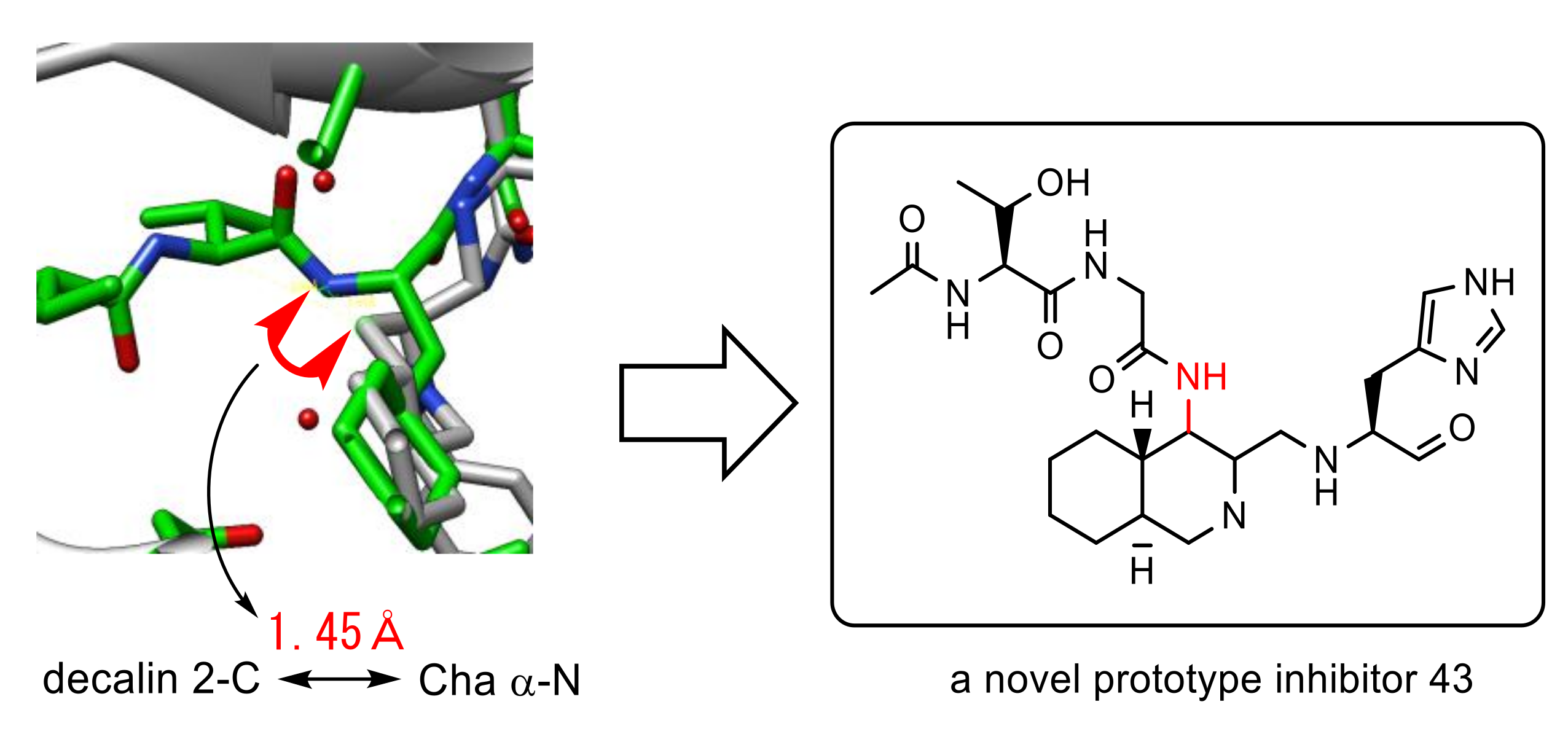
3.6. Rational Design of SARS-CoV 3CLpro Inhibitors Based on Structural Analyses
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of viruses from a high proportion of patients with colds. Lancet 1966, 108, 257. [Google Scholar] [CrossRef]
- Hamre, D.; Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190. [Google Scholar] [CrossRef]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-vanDillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhou, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368. [Google Scholar] [CrossRef]
- Vabret, A.; Mourez, T.; Dina, J.; van der Hoek, L.; Gouarin, S.; Petitjean, J.; Brouard, J.; Freymuth, F. Human coronavirus NL63, France. Emerg. Infect. Dis. 2005, 11, 1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arden, K.E.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. New human coronavirus, HCoV-NL63, associated with severe lower respiratory tract disease in Australia. J. Med. Virol. 2005, 75, 455. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Lau, S.K.; Tsoi, H.W.; Huang, Y.; Poon, R.W.; Chu, C.M.; Lee, R.A.; Luk, W.K.; Wong, G.K.; Wong, B.H.; et al. Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J. Infect. Dis. 2005, 192, 1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, F.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Gunther, S.; Preiser, W.; der Werf, S.V.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolensnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles” heel": Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Mielech, A.M.; Deng, X.; Baker, S. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013, 87, 11955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. COVID-19 Situation Reprorts, 163. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 13 August 2020).
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriage, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, D.; Sun, X.; Curth, U.; Drosten, C.; Sanerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–410. [Google Scholar] [CrossRef] [Green Version]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chem, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordingley, M.G.; Register, R.B.; Callahan, P.L.; Garsky, V.M.; Colonno, R.J. Cleavage of small peptides in vitro by human rhinovirus 14 3C protease expressed in Escherichia coli. J. Virol. 1989, 63, 5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, A.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weibbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Viol. 2003, 84, 2305. [Google Scholar] [CrossRef]
- Shi, J.; Wei, Z.; Song, J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: Defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004, 279, 24765. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef]
- Wang, H.; He, S.; Deng, W.; Zhang, Y.; Li, G.; Sun, J.; Zhao, W.; Guo, Y.; Yin, Z.; Li, D.; et al. Comprehensive Insights into the Catalytic Mechanism of Middle East Respiratory Syndrome 3C-Like Protease and Severe Acute Respiratory Syndrome 3C-Like Protease. Acs. Catal. 2020, 10, 5871. [Google Scholar] [CrossRef]
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV pp1a/ab nsp7-10 region. Biochem. J. 2020, 477, 1009. [Google Scholar] [CrossRef] [Green Version]
- Akaji, K.; Konno, H.; Onozuka, M.; Makino, A.; Saito, H.; Nosaka, K. Evaluation of peptide-aldehyde inhibitors using R188I mutant of SARS 3CL protease as a proteolysis-resistant mutant. Bioorg. Med. Chem. 2008, 16, 9400. [Google Scholar] [CrossRef]
- Jain, R.P.; Pettersson, H.I.; Zhang, J.; Aull, K.D.; Fortin, P.D.; Huitema, C.; Eltis, L.D.; Parrish, J.C.; James, M.N.; Wishart, D.S.; et al. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 2004, 47, 6113. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Xi, K.; Ratia, K.; Santarsiero, B.D.; Fu, W.; Harcourt, B.H.; Rota, P.A.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005, 48, 6767. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gui, C.; Luo, X.; Yang, Q.; Gunther, S.; Scandella, E.; Drosten, C.; Bai, D.; He, X.; Ludewig, B.; et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005, 79, 7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, J.E.; Elowe, N.H.; Huitema, C.; Fortin, P.D.; Cechetto, J.D.; Eltis, L.D.; Brown, E.D. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004, 11, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shie, J.J.; Fang, J.-M.; Kuo, T.-H.; Kuo, C.-J.; Liang, P.-H.; Huang, H.-J.; Wu, Y.-T.; Jan, J.-T.; Cheng, Y.-S.E.; Wong, C.-H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha,beta-unsaturated esters. Bioorg. Med. Chem. 2005, 13, 5240. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, S.-J.; Hsu, M.-F.; Wu, J.-D.; Tseng, C.-T.K.; Liu, Y.-F.; Chen, H.-C.; Kuo, C.-W.; Wu, C.-S.; Chang, L.-W. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006, 49, 4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-C.; Kuo, C.-J.; Ko, T.-P.; Hsu, M.-F.; Tsui, Y.-C.; Chang, S.-C.; Yang, S.; Chen, S.-J.; Chen, H.-C.; Hsu, M.-C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646. [Google Scholar] [CrossRef] [Green Version]
- Akaji, K.; Konno, H.; Mitsui, H.; Teruya, K.; Shimamoto, Y.; Hattori, Y.; Ozaki, T.; Kusunoki, M.; Sanjoh, A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011, 54, 7962. [Google Scholar] [CrossRef]
- Zhang, H.-Z.; Zhang, H.; Kemnitzer, W.; Tseng, B.; Cinatl, J.; Michaelis, M.; Doerr, H.W.; Cai, S.X. Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS-CoV) inhibitors. J. Med. Chem. 2006, 49, 1198. [Google Scholar] [CrossRef]
- Bacha, U.; Barrila, J.; Gabelli, S.B.; Kiso, Y.; Amzel, L.M.; Freire, E. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease. Chem. Biol. Drug Des. 2008, 72, 34. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.-M.; Yang, W.-B.; Kuo, T.-H.; Tsai, K.-C.; Lin, C.-H.; Yang, A.-S.; Liang, P.-H.; Wong, C.-H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. J. Med. Chem. 2008, 16, 4652. [Google Scholar] [CrossRef]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Chen, S.E.; Naser-Tavakolian, A.; et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: Design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372. [Google Scholar] [CrossRef] [PubMed]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Kiso, Y.; Kawasaki, Y.; et al. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: Structure-activity relationship study. Eur. J. Med. Chem. 2013, 65, 436. [Google Scholar] [CrossRef] [PubMed]
- Chuck, C.-P.; Chen, C.; Ke, Z.; Wan, D.C.-C.; Chow, H.-F.; Wong, K.-B. Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases. Eur. J. Med. Chem. 2013, 59, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 2011, 92, 204. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhu, H.-M.; Niu, G.-J.; Shi, E.-Z.; Chen, J.; Sun, B.; Chen, W.-Q.; Zhou, H.-G.; Yang, C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2014, 22, 292. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Ko, J.-A.; Kim, D.W.; Kim, Y.M.; Kwon, H.-J.; Jeong, H.J.; Kim, C.Y.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. J. Enzym. Inhib. Med. Chem. 2016, 31, 23. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Yuk, H.J.; Ryu, H.W.; Lim, S.H.; Kim, K.S.; Park, K.H.; Ryu, Y.B.; Lee, W.S. Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 504. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.H.; Ryu, H.-J.; Lee, S.-H.; Hwang, S.; Breton, V.; Rhee, J.H.; Kim, D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett. 2011, 21, 3088. [Google Scholar] [CrossRef]
- Mukherjee, P.; Desai, P.; Ross, L.; White, E.L.; Avery, M.A. Structure-based virtual screening against SARS-3CL(pro) to identify novel non-peptidic hits. Bioorg. Med. Chem. 2008, 16, 4138. [Google Scholar] [CrossRef]
- Lee, H.; Mittal, A.; Patel, K.; Gatuz, J.L.; Truong, L.; Torres, J.; Mulhearn, D.C.; Johnson, M.E. Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorg. Med. Chem. 2014, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534. [Google Scholar] [PubMed] [Green Version]
- Turlington, M.; Chun, A.; Tomar, S.; Eggler, A.; Grum-Tokars, V.; Jacobs, J.; Daniels, J.S.; Dawson, E.; Saldanha, A.; Chase, P.; et al. Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: Identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett. 2013, 23, 6172. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Wakabayashi, M.; Takanuma, D.; Saito, Y.; Akaji, K. Design and synthesis of a series of serine derivatives as small molecule inhibitors of the SARS coronavirus 3CL protease. Bioorg. Med. Chem. 2016, 24, 1241. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, Y.; Hattori, Y.; Kobayashi, K.; Teruya, K.; Sanjoh, A.; Nakagawa, A.; Yamashita, E.; Akaji, K. Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg. Med. Chem. 2015, 23, 876. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, K.; Hattori, Y.; Kobayashi, K.; Akaji, K. Evaluation of a non-prime site substituent and warheads combined with a decahydroisoquinolin scaffold as a SARS 3CL protease inhibitor. Bioorg. Med. Chem. 2019, 27, 425. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enz. Inhibi. Med. Chem. 2020, 35, 145. [Google Scholar] [CrossRef] [Green Version]
- Vellingiri, B.; Jayaramayya, K.; Iyer, M.; Narayanasamy, A.; Govindasamy, V.; Giridharan, B.; Ganesan, S.; Venugopal, A.; Venkatesan, D.; Ganesan, H.; et al. COVID-19: A promising cure for the global panic. Sci. Total Environ. 2020, 725, 138277. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Neioo, H.M.O.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akaji, K.; Konno, H. Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease. Molecules 2020, 25, 3920. https://doi.org/10.3390/molecules25173920
Akaji K, Konno H. Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease. Molecules. 2020; 25(17):3920. https://doi.org/10.3390/molecules25173920
Chicago/Turabian StyleAkaji, Kenichi, and Hiroyuki Konno. 2020. "Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease" Molecules 25, no. 17: 3920. https://doi.org/10.3390/molecules25173920
APA StyleAkaji, K., & Konno, H. (2020). Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease. Molecules, 25(17), 3920. https://doi.org/10.3390/molecules25173920