
Recent Progress in the Discovery and Development of 2-Nitroimidazooxazines and 6-Nitroimidazooxazoles to Treat Tuberculosis and Neglected Tropical Diseases †


Abstract
:1. Introduction
2. 2-Nitroimidazooxazines
2.1. Pretomanid (PA-824; 2)
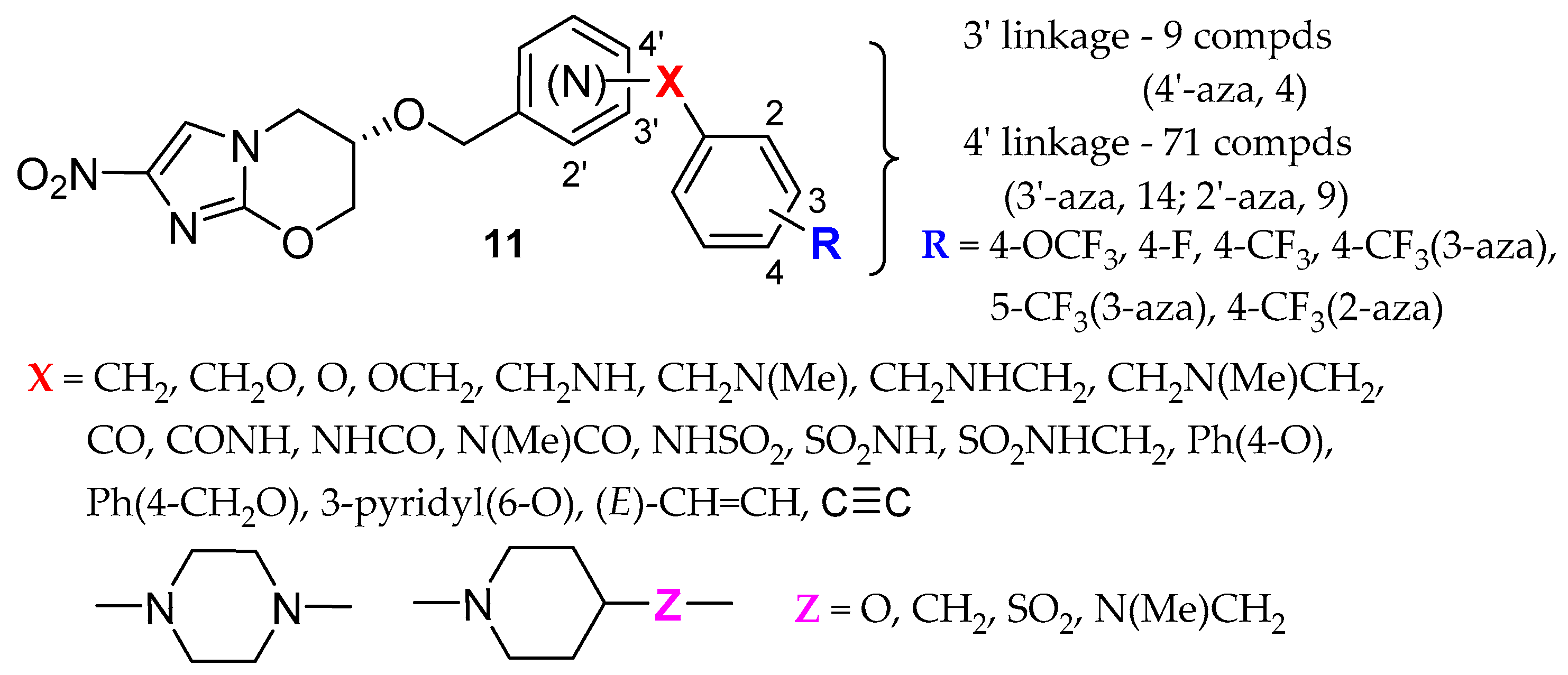
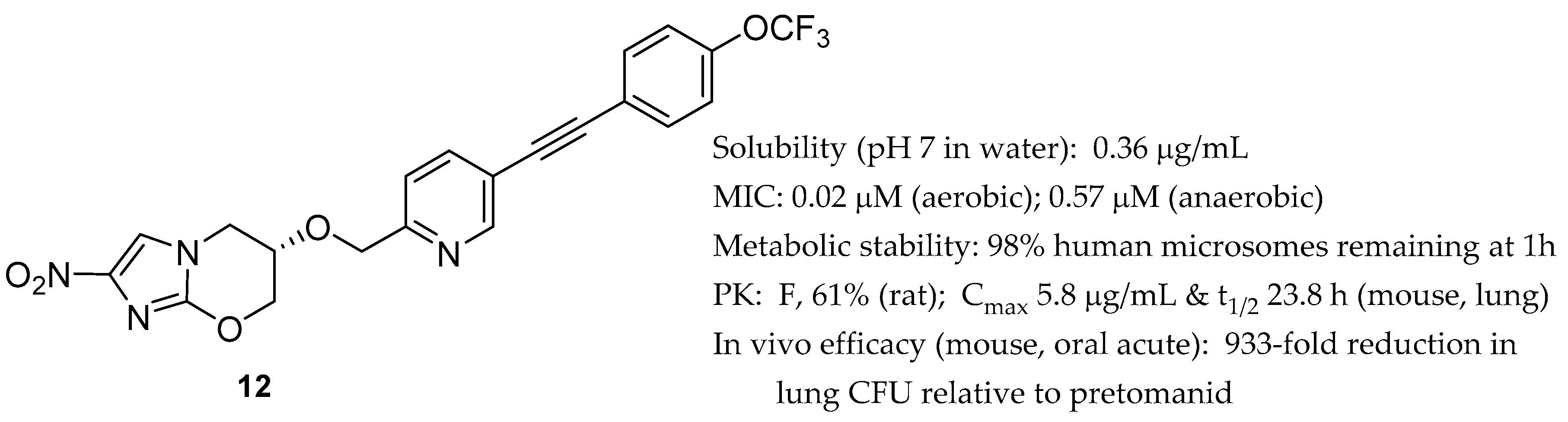
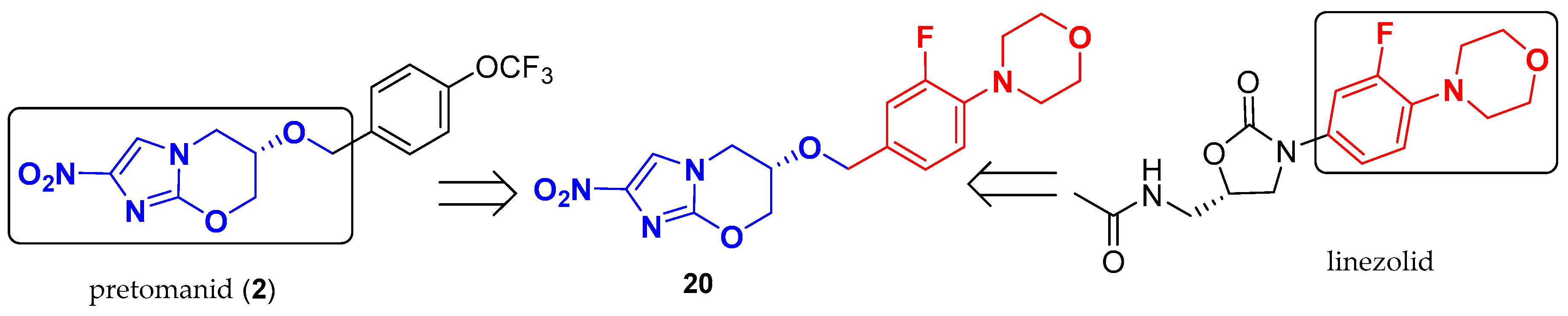
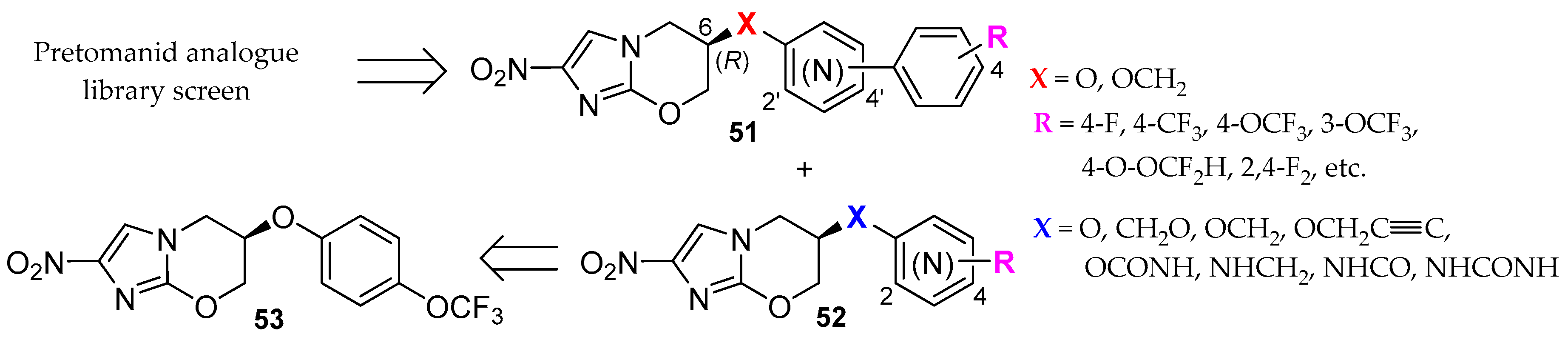
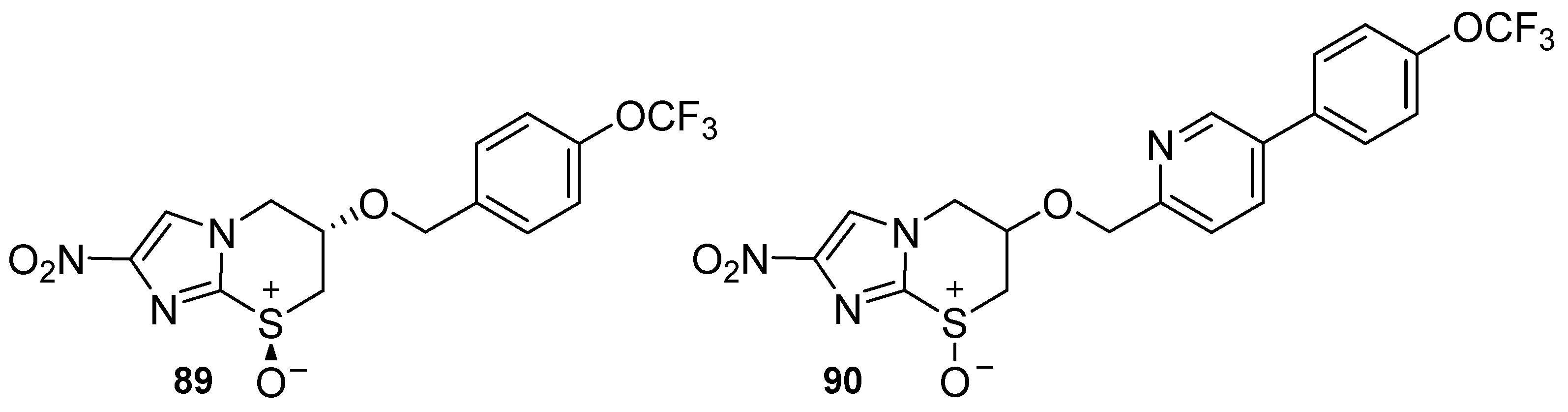
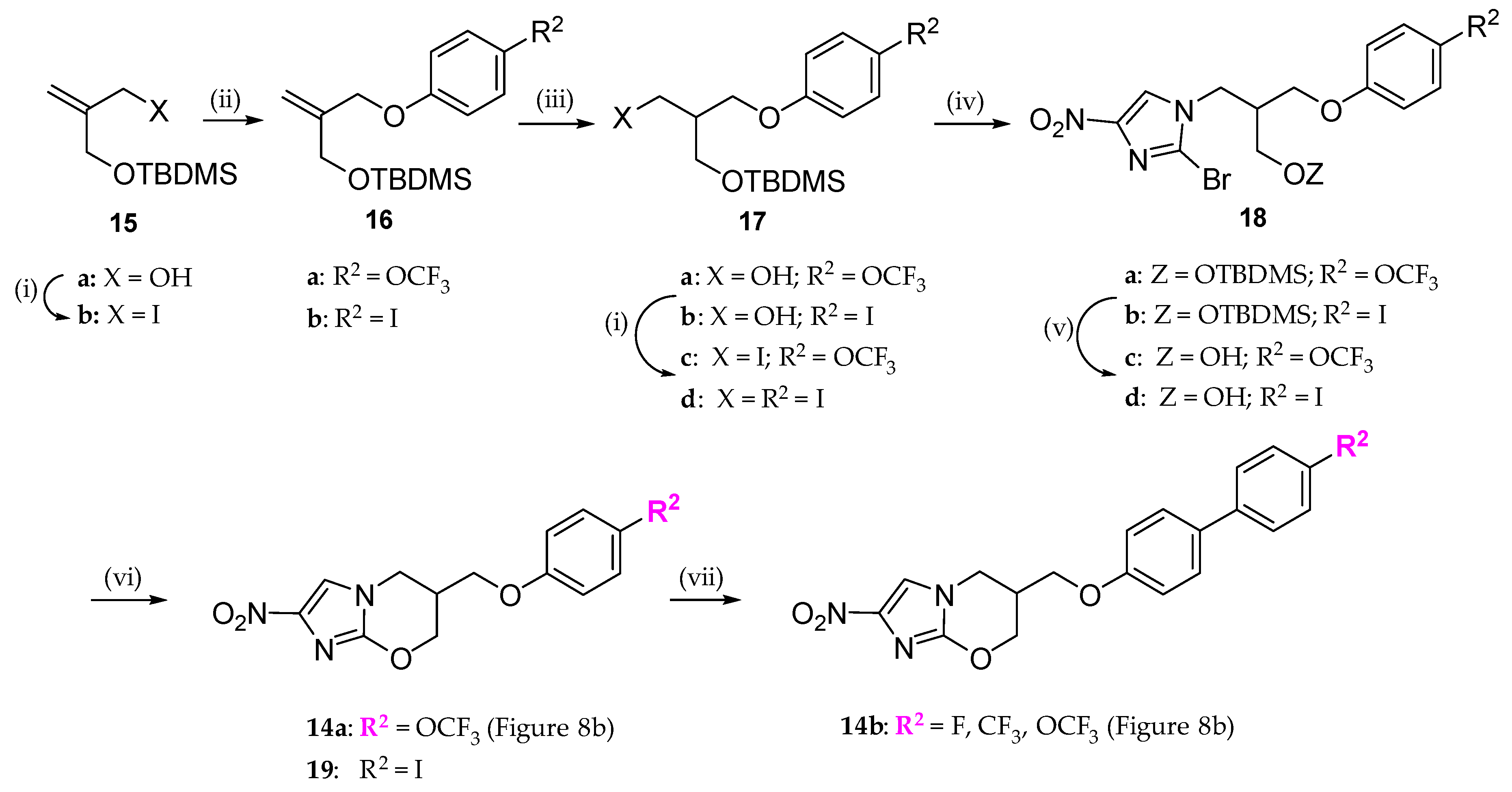
2.2. Pretomanid (2-Nitroimidazooxazine) Analogues
3. 6-Nitroimidazooxazoles
3.1. Delamanid (OPC-67683; 3)
3.2. Delamanid (6-Nitroimidazooxazole) Analogues
4. Scaffold Modifications to Bicylic Nitroimidazoles
5. Computed Properties
6. Syntheses of Clinical Agents
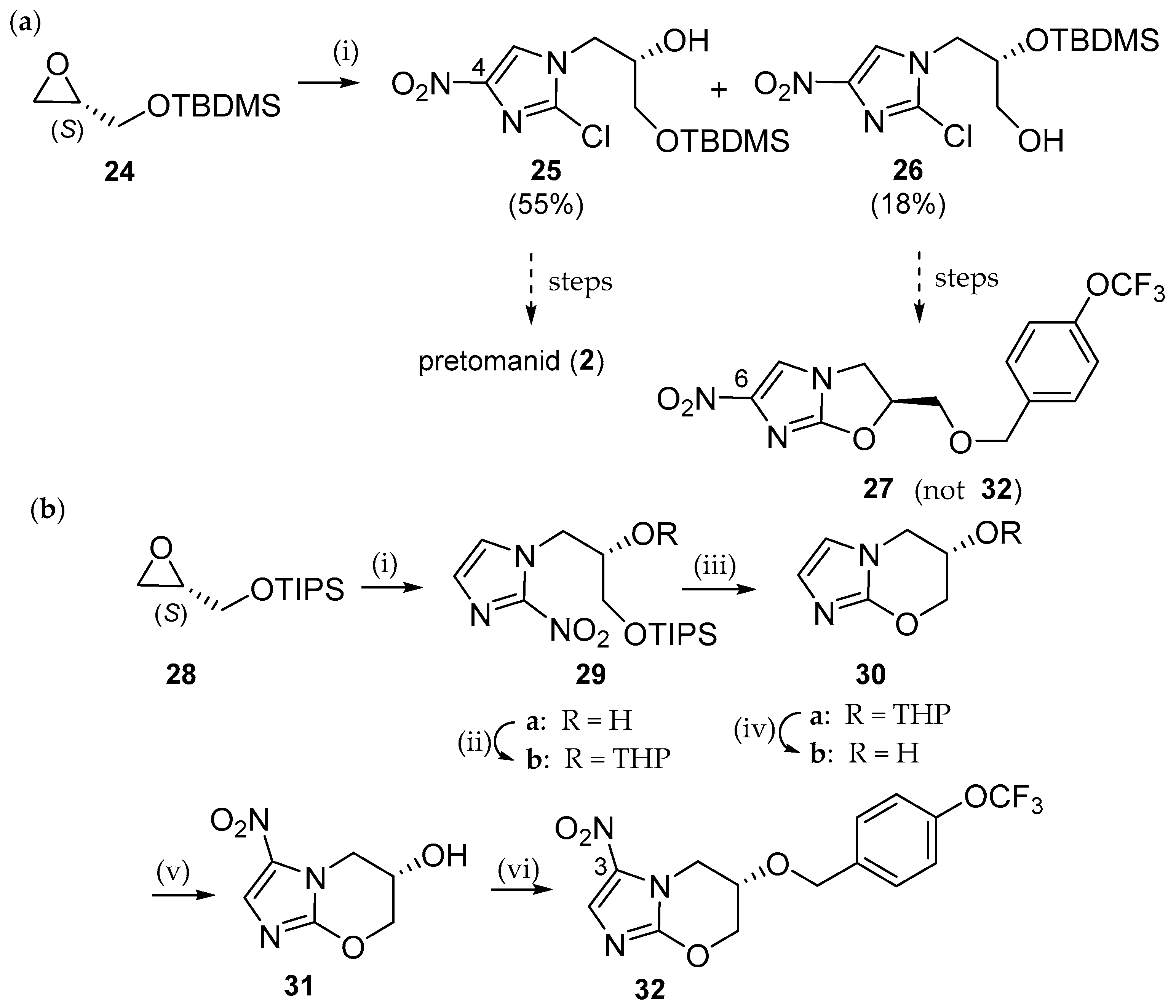
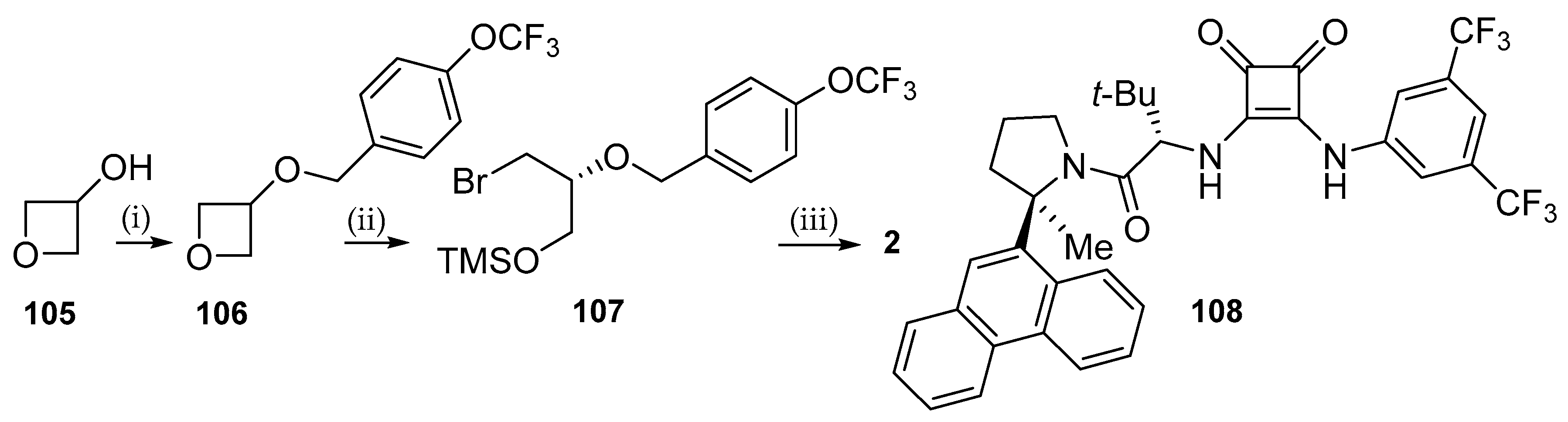
6.1. Pretomanid (PA-824; 2)
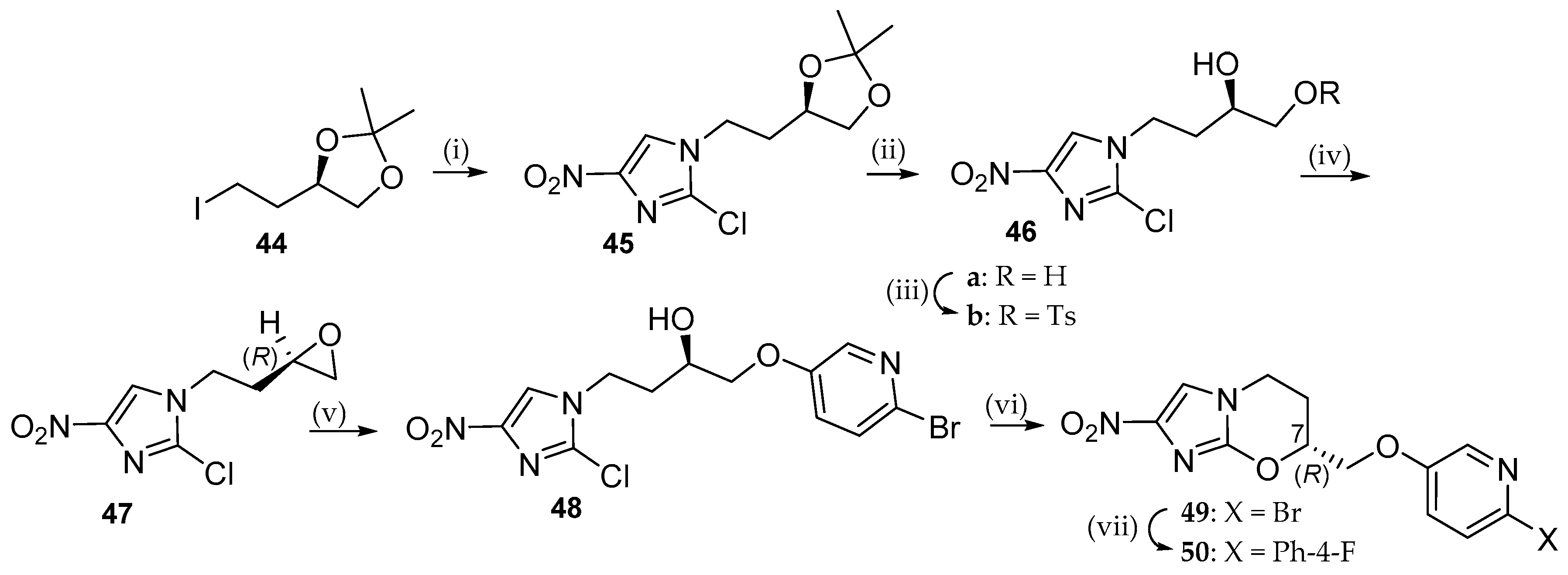
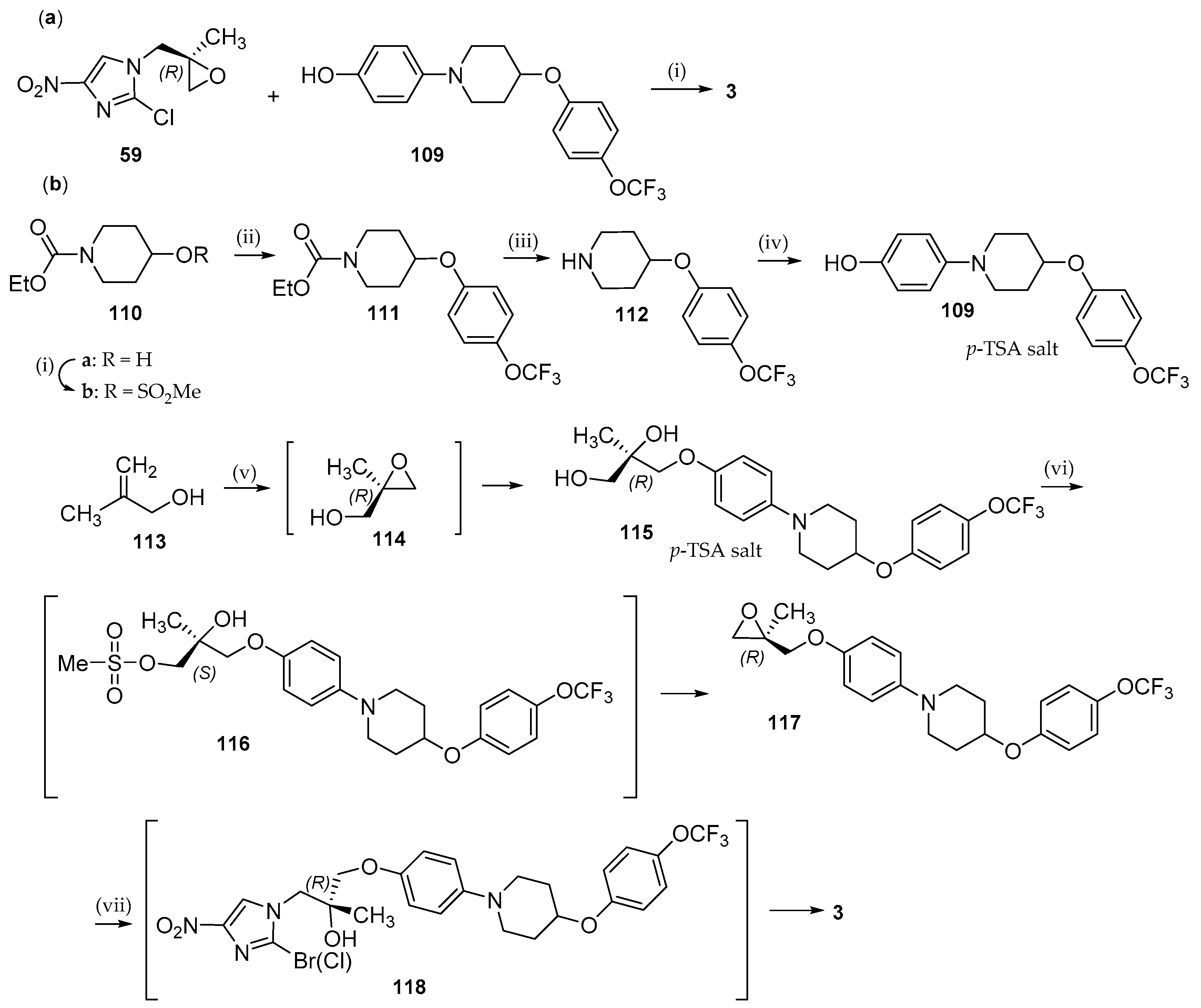
6.2. Delamanid (OPC-67683; 3)
6.3. DNDI-VL-2098 (40)
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland; Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 19 August 2020).
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: From extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2014, 2, 321–338. [Google Scholar] [CrossRef] [Green Version]
- Abreu, P.A.; Medeiros, C.A.; Borges, J.C.; Bernardino, A.M.R.; Rodrigues, C.R.; Castro, H.C. Tuberculosis, an old disease lacking new therapeutic drugs. Curr. Drug Ther. 2013, 8, 86–98. [Google Scholar] [CrossRef]
- Tornheim, J.A.; Dooley, K.E. The Global Landscape of Tuberculosis Therapeutics. Ann. Rev. Med. 2019, 70, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention: Atlanta, Georgia. Available online: https://www.cdc.gov/globalhealth/ntd/diseases/index.html (accessed on 19 August 2020).
- Urbina, J.A. Specific chemotherapy of Chagas disease: Relevance, current limitations and new approaches. Acta Trop. 2010, 115, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M. Chagas disease in the 21st century: A public health success or an emerging threat? Parasite 2014, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Simarro, P.P.; Jannin, J.G.; Diarra, A. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar]
- Kennedy, P.G. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Ferrins, L.; Rahmani, R.; Baell, J.B. Drug discovery and human African trypanosomiasis: A disease less neglected? Future Med. Chem. 2013, 5, 1801–1841. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Phillips, M.A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11, 1255–1274. [Google Scholar] [CrossRef]
- Torreele, E.; Trunz, B.B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazue, G.; Bray, M.A.; Pecoul, B. Fexinidazole—A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 Human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, D. Recent updates and perspectives on leishmaniasis. J. Infect. Dev. Ctries. 2015, 9, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ready, P.D. Epidemiology of visceral leishmaniasis. Clin. Epidemiol. 2014, 6, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, C.W.; Jarrad, A.M.; Cooper, M.A.; Blaskovich, M.A.T. Nitroimidazoles: Molecular fireworks that combat a broad spectrum of infectious diseases. J. Med. Chem. 2017, 60, 7636–7657. [Google Scholar] [CrossRef]
- Nagarajan, K.; Shankar, R.G.; Rajappa, S.; Shenoy, S.J.; Costa-Pereira, R. Nitroimidazoles. XXI. 2,3-Dihydro-6-nitroimidazo[2,1-b]oxazoles with antitubercular activity. Eur. J. Med. Chem. 1989, 24, 631–633. [Google Scholar] [CrossRef]
- Ashtekar, D.R.; Costa-Perira, R.; Nagrajan, K.; Vishvanathan, N.; Bhatt, A.D.; Rittel, W. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993, 37, 183–186. [Google Scholar] [CrossRef] [Green Version]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Barry, C.E. Is the mycobacterial cell wall a hopeless drug target for latent tuberculosis? Drug Discov. Today Dis. Mech. 2006, 3, 237–245. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.M.; Barry, C.E., III. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Manjunatha, U.; Boshoff, H.I.M.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; et al. PA-824 Kills Nonreplicating Mycobacterium tuberculosis by Intracellular NO Release. Science 2008, 322, 1392–1395. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Moraski, G.C.; Krchnak, V.; Miller, P.A.; Colon-Martinez, M.; Herrero, E.; Oliver, A.G.; Miller, M.J. Thiolates Chemically Induce Redox Activation of BTZ043 and Related Potent Nitroaromatic Anti-Tuberculosis Agents. J. Am. Chem. Soc. 2013, 135, 3539–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Roberts, A.J.; Norval, S.; Patterson, S.; Foth, B.J.; Berriman, M.; Read, K.D.; Fairlamb, A.H. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLOS Pathog. 2016, 12, e1005971/1–e1005971/22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, C.E.; Crick, D.C.; McNeil, M.R. Targeting the formation of the cell wall core of M. tuberculosis. Infect. Disord. Drug Targets 2007, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Rivers, E.C.; Mancera, R.L. New anti-tuberculosis drugs with novel mechanisms of action. Curr. Med. Chem. 2008, 15, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Gao, C.; Huang, Z.-G.; Luo, W.; Liu, K.-L.; Peng, C.-T.; Ding, C.Z.; Li, J.; Chen, S.-H.; Yu, L.-T. Discovery and evaluation of novel nitrodihydroimidazooxazoles as promising anti-tuberculosis agents. Bioorg. Med. Chem. Lett. 2019, 29, 2511–2515. [Google Scholar] [CrossRef]
- Yempalla, K.R.; Munagala, G.; Singh, S.; Kour, G.; Sharma, S.; Chib, R.; Kumar, S.; Wazir, P.; Singh, G.D.; Raina, S.; et al. Synthesis and Biological Evaluation of Polar Functionalities Containing Nitrodihydroimidazooxazoles as Anti-TB Agents. ACS Med. Chem. Lett. 2015, 6, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Falzari, K.; Zhu, Z.; Pan, D.; Liu, H.; Hongmanee, P.; Franzblau, S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.H.; Warit, S.; Wan, B.; Hwang, C.H.; Pauli, G.F.; Franzblau, S.G. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Lee, H.S.; Franzblau, S. Microplate alamar blue assay (MABA) and low oxygen recovery assay (lora) for mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 281–292. [Google Scholar] [PubMed]
- Denny, W.A.; Palmer, B.D. The nitroimidazooxazines (PA-824 and analogs): Structure-activity relationship and mechanistic studies. Future Med. Chem. 2010, 2, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Goto, F.; Takemura, N.; Otani, T.; Hasegawa, T.; Tsubouchi, H.; Utsumi, N.; Fujita, S.; Kuroda, H.; Shitsuta, T.; Sasaki, H. 1-Substituted-4-nitroimidazole Compound and Method for Preparing the Same. U.S. Patent 8129544 B2, 6 March 2012. [Google Scholar]
- Thompson, A.M.; Blaser, A.; Anderson, R.F.; Shinde, S.S.; Franzblau, S.G.; Ma, Z.; Denny, W.A.; Palmer, B.D. Synthesis, reduction potentials, and antitubercular activity of ring A/B analogues of the bioreductive drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2009, 52, 637–645. [Google Scholar] [CrossRef]
- Baker, W.R.; Cai, S.; Keeler, E.L. Nitro-[2,1-b]imidazopyran Compounds and Antibacterial Uses Thereof. U.S. Patent 6087358, 11 July 2000. [Google Scholar]
- Barry, C.E., III; Boshoff, H.I.M.; Dowd, C.S. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr. Pharm. Des. 2004, 10, 3239–3262. [Google Scholar] [CrossRef]
- Kim, P.; Zhang, L.; Manjunatha, U.H.; Singh, R.; Patel, S.; Jiricek, J.; Keller, T.H.; Boshoff, H.I.; Barry, C.E., III; Dowd, C.S. Structure-activity relationships of antitubercular nitroimidazoles. 1. Structural features associated with aerobic and anaerobic activities of 4- and 5-Nitroimidazoles. J. Med. Chem. 2009, 52, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.; Kang, S.; Boshoff, H.I.; Jiricek, J.; Collins, M.; Singh, R.; Manjunatha, U.H.; Niyomrattanakit, P.; Zhang, L.; Goodwin, M.; et al. Structure-activity relationships of antitubercular nitroimidazoles. 2. Determinants of aerobic activity and quantitative structure-activity relationships. J. Med. Chem. 2009, 52, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Palmer, B.D.; Thompson, A.M.; Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A. Synthesis and structure-activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 282–294. [Google Scholar] [CrossRef]
- Mukherjee, T.; Boshoff, H. Nitroimidazoles for the treatment of TB: Past, present and future. Future Med. Chem. 2011, 3, 1427–1454. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Manjunatha, U.H.; Goodwin, M.B.; Knox, J.E.; Lipinski, C.A.; Keller, T.H.; Barry, C.E., III; Dowd, C.S. Synthesis and antitubercular activity of 7-(R)- and 7-(S)-methyl-2-nitro-6-(S)-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazines, analogues of PA-824. Bioorg. Med. Chem. Lett. 2008, 18, 2256–2262. [Google Scholar] [CrossRef] [Green Version]
- Bollo, S.; Nunez-Vergara, L.J.; Kang, S.; Zhang, L.; Boshoff, H.I.; Barry, C.E., III; Squella, J.A.; Dowd, C.S. The effect of 5-substitution on the electrochemical behavior and antitubercular activity of PA-824. Bioorg. Med. Chem. Lett. 2011, 21, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Palmer, B.D.; Denny, W.A.; Thompson, A.M. Synthesis and structure-activity relationships of antitubercular 2-Nitroimidazooxazines bearing heterocyclic side chains. J. Med. Chem. 2010, 53, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Kmentova, I.; Sutherland, H.S.; Palmer, B.D.; Blaser, A.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A.; Thompson, A.M. Synthesis and structure-activity relationships of aza- and diazabiphenyl analogues of the antitubercular drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 8421–8439. [Google Scholar] [CrossRef]
- Upton, A.M.; Yang, T.J.; Mdluli, K.; Ma, Z.; Cho, S.; Kim, Y.; Wang, Y.; Franzblau, S.G.; Lu, Y.; Wang, B.; et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Global Alliance for TB Drug Development: New York, NY, USA. Available online: https://www.tballiance.org/news/phase-1-clinical-trial-tb-drug-candidate-tba-354-discontinued (accessed on 19 August 2020).
- Ntshangase, S.; Shobo, A.; Kruger, H.G.; Asperger, A.; Niemeyer, D.; Arvidsson, P.I.; Govender, T.; Baijnath, S. The downfall of TBA-354—A possible explanation for its neurotoxicity via mass spectrometric imaging. Xenobiotica 2018, 48, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Pretomanid: First approval. Drugs 2019, 79, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Cherian, J.; Choi, I.-H.; Nayyar, A.; Manjunatha, U.H.; Mukherjee, T.; Lee, Y.-S.; Boshoff, H.I.; Singh, R.; Ha, Y.-H.; Goodwin, M.; et al. Structure-activity relationships of antitubercular nitroimidazoles. 3. Exploration of the linker and lipophilic tail of ((S)-2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-(4-trifluoromethoxybenzyl)amine (6-Amino PA-824). J. Med. Chem. 2011, 54, 5639–5659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.M.; Sutherland, H.S.; Palmer, B.D.; Kmentova, I.; Blaser, A.; Franzblau, S.G.; Wan, B.-J.; Wang, Y.-H.; Ma, Z.-K.; Denny, W.A. Synthesis and structure-activity relationships of varied ether linker analogues of the antitubercular drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2011, 54, 6563–6585. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tsubouchi, H.; Sasaki, H.; Itotani, M.; Kuroda, H.; Tomishige, T.; Kawasaki, M.; Komatsu, M. Screening for novel antituberculosis agents that are effective against multidrug resistant tuberculosis. Curr. Top. Med. Chem. 2007, 7, 499–507. [Google Scholar] [CrossRef]
- Blaser, A.; Palmer, B.D.; Sutherland, H.S.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Thompson, A.M.; Denny, W.A. Structure-activity relationships for Amide-, Carbamate-, And Urea-Linked analogues of the tuberculosis drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2012, 55, 312–326. [Google Scholar] [CrossRef]
- Brown, A.; Brown, T.B.; Calabrese, A.; Ellis, D.; Puhalo, N.; Ralph, M.; Watson, L. Triazole oxytocin antagonists: Identification of an aryloxyazetidine replacement for a biaryl substituent. Bioorg. Med. Chem. Lett. 2010, 20, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.D.; Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A.; Thompson, A.M. Synthesis and structure-activity relationships for extended side chain analogues of the antitubercular drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2015, 58, 3036–3059. [Google Scholar] [CrossRef]
- Thompson, A.M.; Blaser, A.; Palmer, B.D.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A. Biarylmethoxy 2-nitroimidazooxazine antituberculosis agents: Effects of proximal ring substitution and linker reversal on metabolism and efficacy. Bioorg. Med. Chem. Lett. 2015, 25, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, M.; Mukherjee, T.; Dowd, C.S.; Singh, R.; Niyomrattanakit, P.; Tay, J.A.; Nayyar, A.; Lee, Y.S.; Cherian, J.; Boshoff, H.I.; et al. Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioreductive activation of bicyclic nitroimidazoles. FEBS J. 2012, 279, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Dallimore, J.W.P.; Meo, P. Syntheses of the C(1-6) and C(19-24) Fragments of Lituarines A, B, and C. Org. Lett. 2004, 6, 3857–3859. [Google Scholar] [CrossRef]
- Rakesh; Bruhn, D.F.; Scherman, M.S.; Singh, A.P.; Yang, L.; Liu, J.; Lenaerts, A.J.; Lee, R.E. Synthesis and evaluation of pretomanid (PA-824) oxazolidinone hybrids. Bioorg. Med. Chem. Lett. 2016, 26, 388–391. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B.; et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.M.; Bonnet, M.; Lee, H.H.; Franzblau, S.G.; Wan, B.; Wong, G.S.; Cooper, C.B.; Denny, W.A. Antitubercular Nitroimidazoles revisited: Synthesis and activity of the authentic 3-Nitro isomer of pretomanid. ACS Med. Chem. Lett. 2017, 8, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Haraguchi, Y.; Sasaki, H.; Uematsu, Y.; Tsubouchi, H.; Yata, H.; Shimizu, H.; Kohashi, K.; Itotani, M.; Tai, K.; et al. 6,7-Dihydroimidazo[2,1-b][1,3]oxazine Bactericides. U.S. Patent 9051333 B2, 9 June 2015. [Google Scholar]
- Kang, Y.-G.; Park, C.-Y.; Shin, H.; Singh, R.; Arora, G.; Yu, C.-m.; Lee, I.Y. Synthesis and anti-tubercular activity of 2-nitroimidazooxazines with modification at the C-7 position as PA-824 analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3650–3653. [Google Scholar] [CrossRef]
- Moune, S.; Niel, G.; Busquet, M.; Eggleston, I.; Jouin, P. Total synthesis of dolatrienoic acid: A subunit of dolastatin 14. J. Org. Chem. 1997, 62, 3332–3339. [Google Scholar] [CrossRef]
- Sun, H.; Tawa, G.; Wallqvist, A. Classification of scaffold-hopping approaches. Drug Discov. Today 2012, 17, 310–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.M.; O’Connor, P.D.; Marshall, A.J.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Braillard, S.; Chatelain, E.; Franzblau, S.G.; et al. 7-Substituted 2-Nitro-5,6-dihydroimidazo[2,1-b][1,3]oxazines: Novel antitubercular agents lead to a new preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2017, 60, 4212–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnant, G.-J.; Croft, S.L.; de la Flor, R.; Alavijeh, M.; Yardley, V.; Braillard, S.; Mowbray, C.; Van Bocxlaer, K. Pharmacokinetics and pharmacodynamics of the nitroimidazole DNDI-0690 in mouse models of cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2019, 63, e00829-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs for Neglected Diseases Initiative: Geneva, Switzerland. Available online: https://www.dndi.org/diseases-projects/portfolio/dndi-0690 (accessed on 19 August 2020).
- Kumar, C.R.; Tsai, C.-H.; Chao, Y.-S.; Lee, J.-C. The first total synthesis of cytopiloyne, an anti-diabetic, polyacetylenic glucoside. Chem. Eur. J. 2011, 17, 8696–8703. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; O’Connor, P.D.; Marshall, A.J.; Blaser, A.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Braillard, S.; Chatelain, E.; et al. Development of (6R)-2-Nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): A new lead for visceral leishmaniasis. J. Med. Chem. 2018, 61, 2329–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiazza, C.; Billard, T.; Dickson, C.; Tlili, A.; Gampe, C.M. Chalcogen OCF3 Isosteres modulate drug properties without introducing inherent liabilities. ChemMedChem 2019, 14, 1586–1589. [Google Scholar] [CrossRef]
- Jiricek, J.; Patel, S.; Keller, T.H.; Barry, C.E.; Dowd, C.S. Nitroimidazole Compounds. U.S. Patent 2008/0275035 A1, 6 November 2007. [Google Scholar]
- Sasaki, H.; Haraguchi, Y.; Itotani, M.; Kuroda, H.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Matsumoto, M.; Komatsu, M.; Tsubouchi, H. Synthesis and antituberculosis activity of a novel series of optically active 6-Nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J. Med. Chem. 2006, 49, 7854–7860. [Google Scholar] [CrossRef]
- Tsubouchi, H.; Sasaki, H.; Kuroda, H.; Itotani, M.; Hasegawa, T.; Haraguchi, Y.; Kuroda, T.; Matsuzaki, T.; Tai, K.; Komatsu, M.; et al. 2,3-Dihydro-6-nitroimidazo[2,1-b]oxazoles. U.S. Patent 7262212 B2, 28 August 2007. [Google Scholar]
- Tsubouchi, H.; Sasaki, H.; Ishikawa, H.; Matsumoto, M. Discovery of delamanid for the treatment of multidrug-resistant pulmonary tuberculosis. In Successful Drug Discovery; Fisher, J., Childers, W.E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; Volume 1, pp. 137–161. [Google Scholar]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef]
- Liu, Y.; Matsumoto, M.; Ishida, H.; Ohguro, K.; Yoshitake, M.; Gupta, R.; Geiter, L.; Hafkin, J. Delamanid: From discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Tsubouchi, H.; Sasaki, H.; Itotani, M.; Haraguchi, Y.; Miyamura, S.; Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Ohguro, K.; et al. 2,3-Dihydro-6-nitroimidazo[2,1-b]oxazoles for the Treatment of Tuberculosis. U.S. Patent 8163753 B2, 24 April 2012. [Google Scholar]
- Singh, P.P.; Munagala, G.; Kushalava, R.Y.; Khan, I.A.; Kalia, N.P.; Rajput, V.S.; Nargotra, A.; Sawant, S.D.; Vishwakarma, R.A. 6-Nitro-2,3-dihydroimidazo[2,1-b]oxazoles and a Process for the Preparation Thereof. U.S. Patent 9845330 B2, 19 December 2017. [Google Scholar]
- Munagala, G.; Yempalla, K.R.; Singh, S.; Sharma, S.; Kalia, N.P.; Rajput, V.S.; Kumar, S.; Sawant, S.D.; Khan, I.A.; Vishwakarma, R.A.; et al. Synthesis of new generation triazolyl- and isoxazolyl-containing 6-nitro-2,3-dihydroimidazooxazoles as anti-TB agents: In vitro, structure-activity relationship, pharmacokinetics and in vivo evaluation. Org. Biomol. Chem. 2015, 13, 3610–3624. [Google Scholar] [CrossRef]
- Yempalla, K.R.; Munagala, G.; Singh, S.; Sharma, S.; Khan, I.A.; Vishwakarma, R.A.; Singh, P.P. Substituted 1,2,3-Triazol-1-yl-methyl-2,3-dihydro-2-methyl-6-nitroimidazo[2,1-b]oxazoles as Anti-Mycobacterial Agents and a Process for the Preparation Thereof. U.S. Patent 9822126 B1, 21 November 2017. [Google Scholar]
- Gupta, S.; Yardley, V.; Vishwakarma, P.; Shivahare, R.; Sharma, B.; Launay, D.; Martin, D.; Puri, S.K. Nitroimidazo-oxazole compound DNDI-VL-2098: An orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. J. Antimicrob. Chemother. 2015, 70, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.M.; O’Connor, P.D.; Blaser, A.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Martin, D.; Franzblau, S.G.; Wan, B.; et al. Repositioning antitubercular 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: Structure-activity studies on a preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2016, 59, 2530–2550. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Hashimoto, Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Ding, C.Z.; Huang, Z.; Chen, S. Anti-pulmonary Tuberculosis Nitroimidazole Derivative. U.S. Patent 10227362 B2, 12 March 2019. [Google Scholar]
- Ding, C.Z.; Huang, Z.; Luo, W.; Chen, S. Anti-pulmonary Tuberculosis Nitroimidazole Derivative. U.S. Patent WO 2019/128963 A1, 4 July 2019. [Google Scholar]
- Mathias, F.; Cohen, A.; Kabri, Y.; Negrao, N.W.; Crozet, M.D.; Docampo, R.; Azas, N.; Vanelle, P. Synthesis and in-vitro evaluation of new 5-substituted 6-nitroimidazooxazoles as antikinetoplastid agents. Eur. J. Med. Chem. 2020, 191, 112146. [Google Scholar] [CrossRef] [PubMed]
- Zaprutko, L.; Zwawiak, J.; Augustynowicz-Kopec, E.; Zwolska, Z.; Bartoszak-Adamska, E.; Nowicki, W. Synthesis, structure and biological evaluation of novel bicyclic nitroimidazole derivatives. Arch. Pharm. 2012, 345, 463–467. [Google Scholar] [CrossRef]
- Zaprutko, L.; Gajdzinski, M.; Michalska, W.; Pietkiewicz, K.; Lutomski, K.; Lukaszewski, Z.; Wrzeciono, U. Azoles. Part 27: Nitroimidazole derivatives: Their antibacterial and fungicidal activity and electron affinity. Pharmazie 1989, 44, 817–820. [Google Scholar] [CrossRef]
- Musonda, C.C.; Edlin, C.D.; Boyle, G.A. Nitroimidazoxadiazocine Compounds. WO 2013/072903 A1, 23 May 2013. [Google Scholar]
- Thompson, A.M.; Blaser, A.; Palmer, B.D.; Anderson, R.F.; Shinde, S.S.; Launay, D.; Chatelain, E.; Maes, L.; Franzblau, S.G.; Wan, B.; et al. 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]thiazoles: Facile synthesis and comparative appraisal against tuberculosis and neglected tropical diseases. Bioorg. Med. Chem. Lett. 2017, 27, 2583–2589. [Google Scholar] [CrossRef]
- Thompson, A.M.; Marshall, A.J.; Maes, L.; Yarlett, N.; Bacchi, C.J.; Gaukel, E.; Wring, S.A.; Launay, D.; Braillard, S.; Chatelain, E.; et al. Assessment of a pretomanid analogue library for African trypanosomiasis: Hit-to-lead studies on 6-substituted 2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine 8-oxides. Bioorg. Med. Chem. Lett. 2018, 28, 207–213. [Google Scholar] [CrossRef]
- Hill, A.P.; Young, R.J. Getting physical in drug discovery: A contemporary perspective on solubility and hydrophobicity. Drug Discov. Today 2010, 15, 648–655. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Orita, A.; Miwa, K.; Uehara, G.; Otera, J. Integration of solventless reaction in a multi-step process: Application to an efficient synthesis of PA-824. Adv. Synth. Catal. 2007, 349, 2136–2144. [Google Scholar] [CrossRef]
- Marsini, M.A.; Reider, P.J.; Sorensen, E.J. A concise and convergent synthesis of PA-824. J. Org. Chem. 2010, 75, 7479–7482. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.P.; Reddy, K.R.; Reddy, A.V.N.; Krishna, B.V. An Improved Process for Preparing Pretomanid. IN 2016/41030408 A, 9 March 2018. [Google Scholar]
- Rao, D.R.; Malhotra, G.; Pullela, V.S.; Patil, S.L.; Rajeshirke, R.R. Process for the Preparation of Nitroimidazole Compounds, Especially the Tuberculostatic Pretomanid. IN 2016/21026053 A, 2 February 2018. [Google Scholar]
- Chen, G.; Zhu, M.; Chen, Y.; Miao, X.; Guo, M.; Jiang, N.; Zhai, X. An efficient and practical protocol for the production of pretomanid (PA-824) via a novel synthetic strategy. Chem. Pap. 2020, 74, 3937–3945. [Google Scholar] [CrossRef]
- Strassfeld, D.A.; Wickens, Z.K.; Picazo, E.; Jacobsen, E.N. Highly enantioselective, hydrogen-bond-donor catalyzed additions to Oxetanes. J. Am. Chem. Soc. 2020, 142, 9175–9180. [Google Scholar] [CrossRef]
- Flick, A.C.; Ding, H.X.; Leverett, C.A.; Kyne, R.E., Jr.; Liu, K.K.C.; Fink, S.J.; O’Donnell, C.J. Synthetic approaches to the 2014 new drugs. Bioorg. Med. Chem. 2016, 24, 1937–1980. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shinhama, K.; Fujita, N.; Aki, S.; Ogasawara, S.; Utsumi, N. Synthetic Intermediate of Oxazole Compound and Method for Producing the Same. U.S. Patent 8598358 B2, 3 December 2013. [Google Scholar]
- Miyake, M.; Asahina, A.; Okada, T. Method for Producing 1-(4-Hydroxyphenyl)-4-(4-trifluoromethoxyphenoxy)piperidine or Salt Thereof. U.S. Patent 10252995 B2, 9 April 2019. [Google Scholar]
- Tsubouchi, H.; Haraguchi, Y.; Hayakawa, S.; Utsumi, N.; Taira, S.; Tanada, Y.; Fujita, N.; Shinhama, K.; Annaka, K.; Furuta, T. Epoxy Compound and Method for Manufacturing the Same. U.S. Patent 8552188 B2, 8 October 2013. [Google Scholar]
- Zhang, Z.; Li, J.; Wang, G.; Chen, H.; Sun, Y. Synthesis of delamanid. Zhongguo Yiyao Gongye Zazhi 2016, 47, 256–260. [Google Scholar]
- Satam, V.S.; Pedada, S.R.; Kamaraj, P.; Antao, N.; Singh, A.; Hindupur, R.M.; Pati, H.N.; Thompson, A.M.; Launay, D.; Martin, D. Development of a scalable process for the synthesis of DNDI-VL-2098: A potential preclinical drug candidate for the treatment of visceral leishmaniasis. Org. Process. Res. Dev. 2017, 21, 52–59. [Google Scholar] [CrossRef]
- Pedada, S.R.; Satam, V.S.; Tambade, P.J.; Kandadai, S.A.; Hindupur, R.M.; Pati, H.N.; Launay, D.; Martin, D. An improved kilogram-scale synthesis of 2-Bromo-4-nitro-1H-imidazole: A key building block of nitroimidazole drugs. Org. Process. Res. Dev. 2013, 17, 1149–1155. [Google Scholar] [CrossRef]
- Cellitti, S.E.; Shaffer, J.; Jones, D.H.; Mukherjee, T.; Gurumurthy, M.; Bursulaya, B.; Boshoff, H.I.; Choi, I.; Nayyar, A.; Lee, Y.S.; et al. Structure of Ddn, the deazaflavin-dependent nitroreductase from mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 2012, 20, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, J.R. New developments in crystallography: Exploring its technology, methods and scope in the molecular biosciences. Biosci. Rep. 2017, 37, BSR20170204. [Google Scholar] [CrossRef] [PubMed] [Green Version]







































{kind=link}
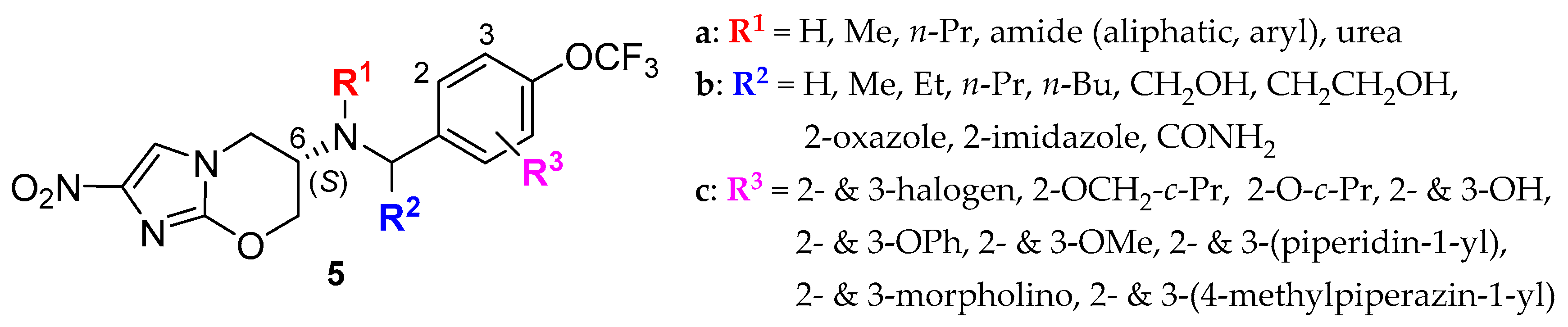{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Structure | MIC M.tb. a | MW | TPSA (Å) b | cLogP b | cLogD7.4 b | SFI c | Aq. Sol. |
|---|---|---|---|---|---|---|---|---|
| 2 |  | 0.05 | 359.26 | 88.65 | 4.50 | 4.14 | 6.14 | 19 [47] d 53 µM |
| 3 |  | 0.01 | 534.49 | 101.12 | 6.46 | 6.14 | 9.14 | 0.31 [85] d 0.6 µM |
| 4 |  | 0.05 | 436.35 | 101.54 | 5.27 | 4.95 | 7.95 | 2.3 [47] d 5.3 µM |
| 10 |  | 0.03 | 457.37 | 112.20 | 4.75 | 4.05 | 6.05 | 17 [55] d 37 µM |
| 12 |  | 0.02 | 460.37 | 101.54 | 5.32 | 5.13 | 8.13 | 0.36 [57] d 0.8 µM |
| 40 |  | 0.05 | 357.29 | 79.42 | 4.72 | 4.19 | 6.19 | 2.4 [85] d 6.7 µM |
| 50 |  | 0.11 | 370.34 | 92.31 | 3.78 | 3.69 | 6.69 | 0.45 [68] d 1.2 µM |
| 53 |  | 31 | 345.23 | 88.65 | 4.50 | 4.10 | 6.10 | 12 [72] d 35 µM |
| rifampin | 0.03 | 822.95 | 220.15 | 3.76 | 2.75 | 2.75 | 2.5 mg/mL e 3.0 mM | |
| bedaquiline | 0.04 | 554.53 | 32.70 | 7.24 | 5.82 | 10.82 | <0.06 µg/mL [97] f <0.1 µM | |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Showalter, H.D. Recent Progress in the Discovery and Development of 2-Nitroimidazooxazines and 6-Nitroimidazooxazoles to Treat Tuberculosis and Neglected Tropical Diseases. Molecules 2020, 25, 4137. https://doi.org/10.3390/molecules25184137
Showalter HD. Recent Progress in the Discovery and Development of 2-Nitroimidazooxazines and 6-Nitroimidazooxazoles to Treat Tuberculosis and Neglected Tropical Diseases. Molecules. 2020; 25(18):4137. https://doi.org/10.3390/molecules25184137
Chicago/Turabian StyleShowalter, Hollis D. 2020. "Recent Progress in the Discovery and Development of 2-Nitroimidazooxazines and 6-Nitroimidazooxazoles to Treat Tuberculosis and Neglected Tropical Diseases" Molecules 25, no. 18: 4137. https://doi.org/10.3390/molecules25184137
APA StyleShowalter, H. D. (2020). Recent Progress in the Discovery and Development of 2-Nitroimidazooxazines and 6-Nitroimidazooxazoles to Treat Tuberculosis and Neglected Tropical Diseases. Molecules, 25(18), 4137. https://doi.org/10.3390/molecules25184137