
Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of Y2R Cysteine Mutants
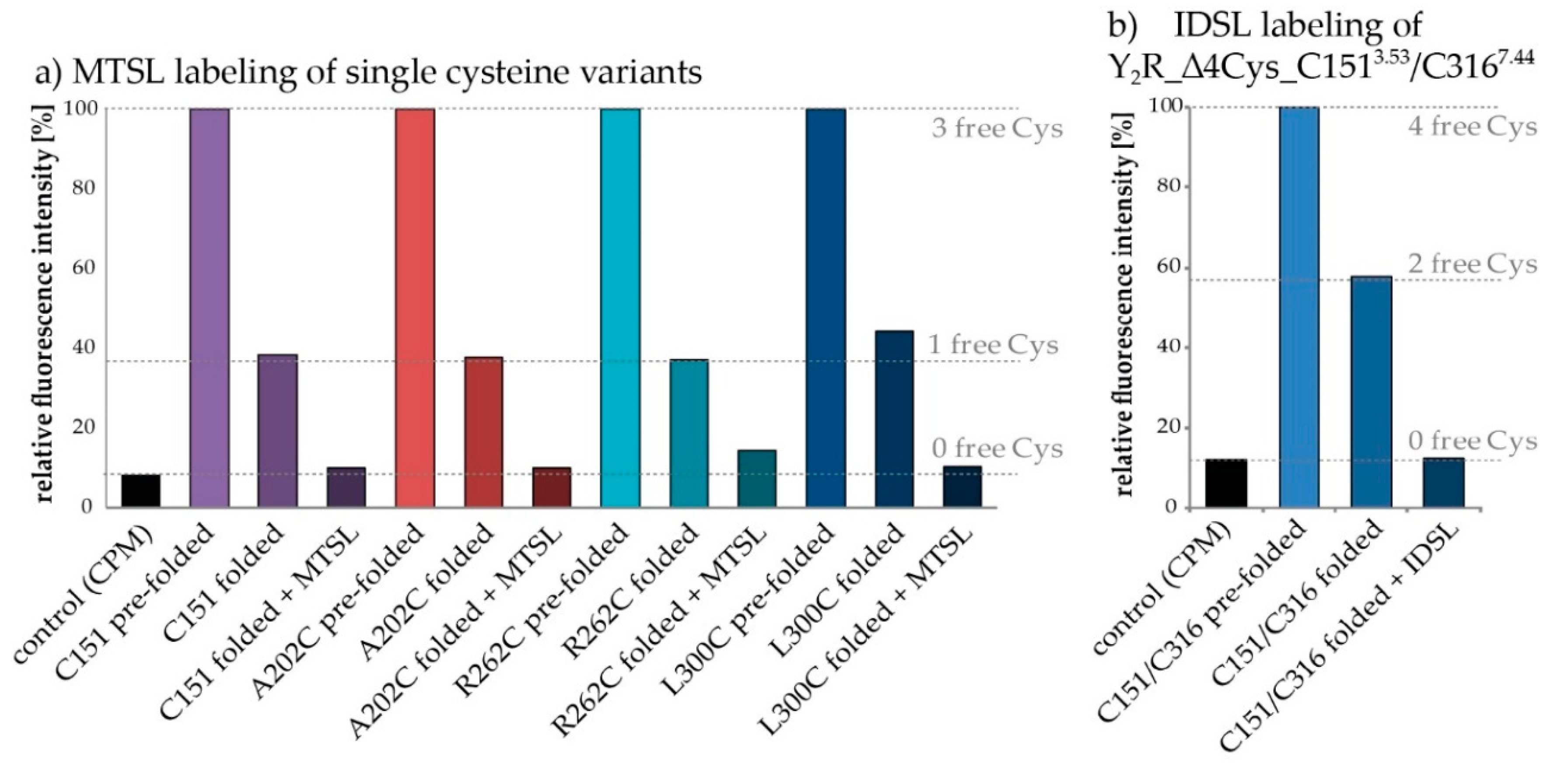
2.2. Protein Expression, Membrane Reconstitution and Spin-Labeling of Y2R Cysteine Mutants
2.3. CW-EPR Measurement Analysis of Y2R Single Cysteine Mutants
2.3.1. CW-EPR of Extracellular Positions A2025.21C and L3007.28C
2.3.2. CW-EPR of Intracellular Positions R2626.29C and C1513.53
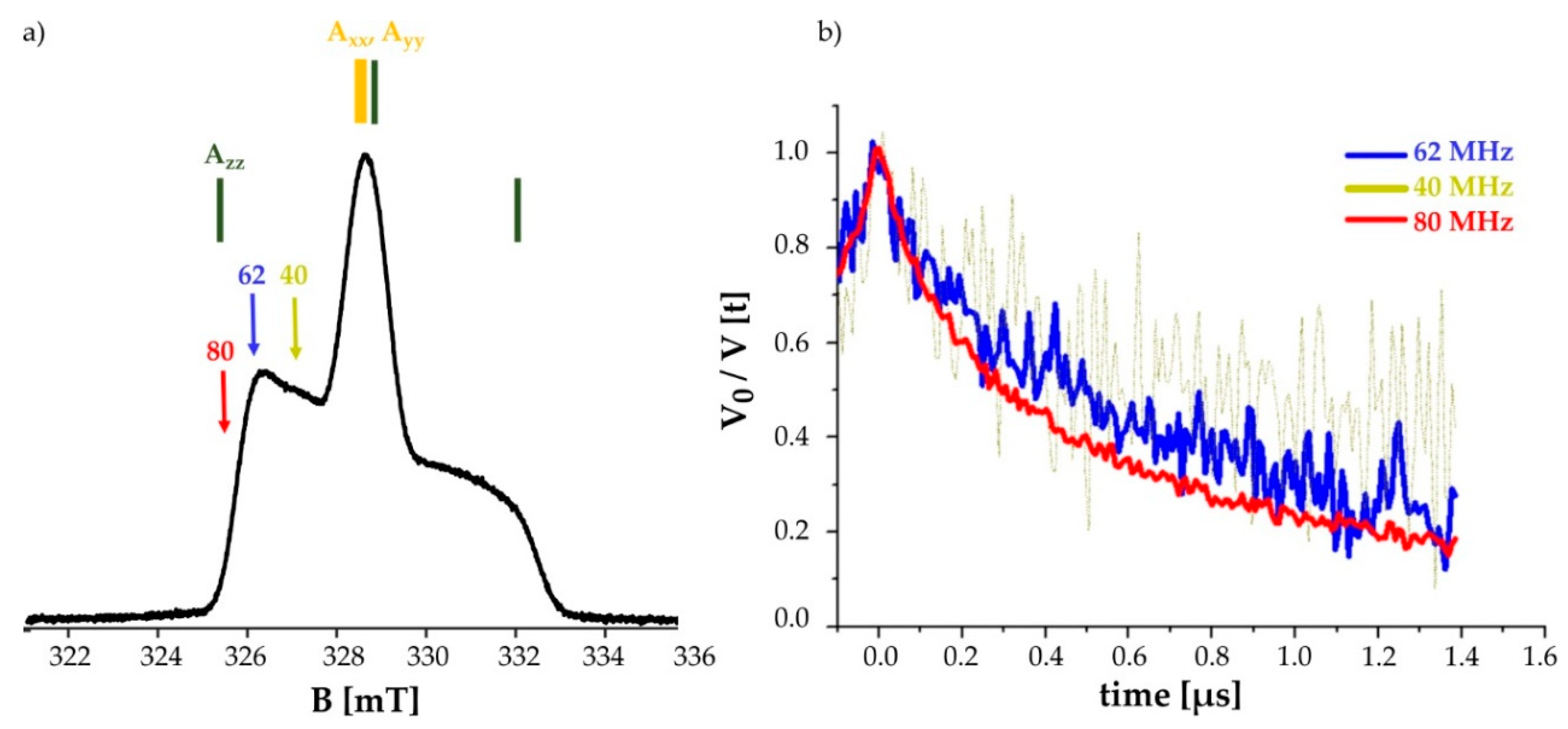
2.3.3. CW-EPR of Transmembrane Position C3167.44
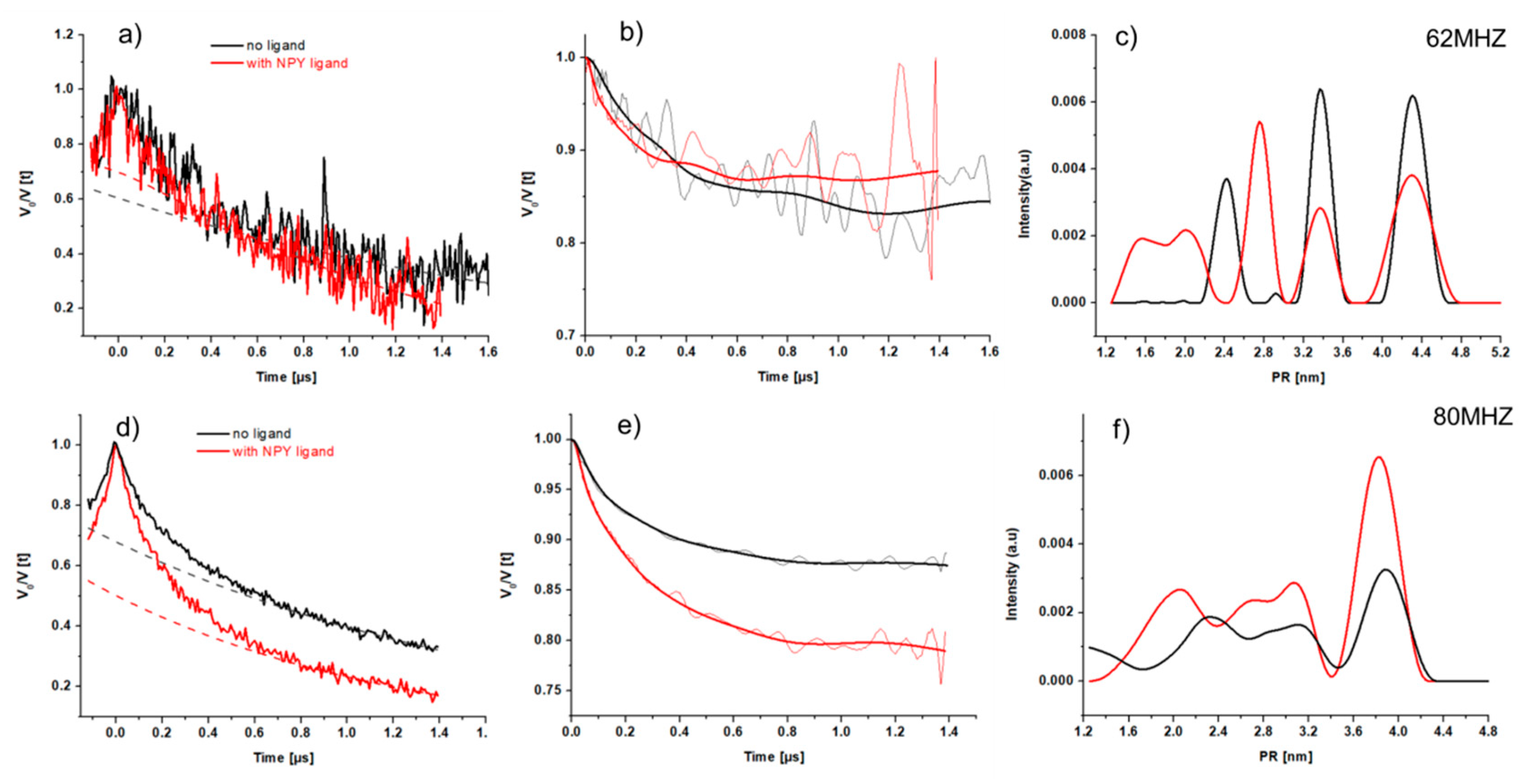
2.3.4. CW-EPR of Single Mutants in the Presence of NPY
2.3.5. CW-EPR of Double Mutants C1513.53/C3167.44
2.4. Pulsed EPR Measurement on the Double Mutated C1513.53/C3167.44
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Peptide Synthesis
4.3. Generation of Plasmids
4.4. Cell Culture Studies
4.4.1. Live-Cell Fluorescence Microscopy
4.4.2. Signal Transduction via Chimeric GαΔ6qi4myr
4.4.3. Signal Transduction via Endogenous Gαi/o
4.4.4. Radioligand Binding Assays
4.5. Y2R Expression and Purification
4.6. In Vitro Folding and Nitroxide Spin Labeling of the Y2R-Variants
4.7. Flourescence Spectroscopy
4.8. EPR-Spectroscopy
4.8.1. CW-EPR Spectroscopy
4.8.2. DEER Experiments
4.9. Analysis Tools
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Capper, M.J.; Wacker, D. How the ubiquitous GPCR receptor family selectively activates signalling pathways. Nature 2018, 558, 529–530. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.; Schertler, G.F. New G-protein-coupled receptor crystal structures: Insights and limitations. Trends Pharmacol. Sci. 2008, 29, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, B.; Feng, D.; Hu, H.; Chu, M.; Qu, Q.; Tarrasch, J.T.; Li, S.; Sun, K.T.; Kobilka, B.K.; et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546, 248–253. [Google Scholar] [CrossRef]
- Liang, Y.L.; Khoshouei, M.; Radjainia, M.; Zhang, Y.; Glukhova, A.; Tarrasch, J.; Thal, D.M.; Furness, S.G.B.; Christopoulos, G.; Coudrat, T.; et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 2017, 546, 118–123. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C. Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 2017, 56, 5628–5634. [Google Scholar] [CrossRef]
- Horst, R.; Liu, J.J.; Stevens, R.C.; Wuthrich, K. beta(2)-adrenergic receptor activation by agonists studied with (1)(9)F NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 10762–10765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the mu-Opioid Receptor. Angew. Chem. Int. Ed. Engl. 2015, 54, 15771–15776. [Google Scholar] [CrossRef] [Green Version]
- Solt, A.S.; Bostock, M.J.; Shrestha, B.; Kumar, P.; Warne, T.; Tate, C.G.; Nietlispach, D. Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat. Commun. 2017, 8, 1795. [Google Scholar] [CrossRef]
- Krug, U.; Gloge, A.; Schmidt, P.; Becker-Baldus, J.; Bernhard, F.; Kaiser, A.; Montag, C.; Gauglitz, M.; Vishnivetskiy, S.A.; Gurevich, V.V.; et al. The Conformational Equilibrium of the Neuropeptide Y2 Receptor in Bilayer Membranes. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, W.L.; Altenbach, C.; Hubbell, C.M.; Khorana, H.G. Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 2003, 63, 243–290. [Google Scholar] [PubMed]
- Steinhoff, H.J. A simple method for determination of rotational correlation times and separation of rotational and polarity effects from EPR spectra of spin-labeled biomolecules in a wide correlation time range. J. Biochem. Biophys. Methods 1988, 17, 237–247. [Google Scholar] [CrossRef]
- Hubbell, W.L.; Cafiso, D.S.; Altenbach, C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000, 7, 735–739. [Google Scholar] [CrossRef]
- Jeschke, G. DEER distance measurements on proteins. Ann. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef] [Green Version]
- Hinderberger, D.; Jeschke, G. Site-Specific Characterization of Structure and Dynamics of Complex Materials by EPR Spin Probes. Mod. Magn. Reson. 2006, 1529–1537. [Google Scholar] [CrossRef]
- Schiemann, O.; Prisner, T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef]
- Toledo, W.D.; Khramtsov, V.V.; Cascio, D.; Altenbach, C.; Hubbell, W.L. Structure and dynamics of an imidazoline nitroxide side chain with strongly hindered internal motion in proteins. J. Magn. Reson. 2013, 232, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.E.; Pannier, M.; Diederich, F.; Gramlich, V.; Hubrich, M.; Spiess, H.W. Determination of End-to-End Distances in a Series of TEMPO Diradicals of up to 2.8 nm Length with a New Four-Pulse Double Electron Electron Resonance Experiment. Angew. Chem. Int. Ed. Engl. 1998, 37, 2833–2837. [Google Scholar] [CrossRef]
- Junk, M.J.; Spiess, H.W.; Hinderberger, D. DEER in biological multispin-systems: A case study on the fatty acid binding to human serum albumin. J. Magn. Reson. 2011, 210, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Milov, A.D.; Salikhov, K.M.; Shirov, M.D. Application of ELDOR in electron-spin echo for paramagnetic center space distribution in solids. Fiz. Tverd. Tela 1981, 23, 975–982. [Google Scholar]
- Schiemann, O.; Weber, A.; Edwards, T.E.; Prisner, T.F.; Sigurdsson, S.T. Nanometer distance measurements on RNA using PELDOR. J. Am. Chem. Soc. 2003, 125, 3434–3435. [Google Scholar] [CrossRef] [PubMed]
- Haeri, H.H.; Blaffert, J.; Schöffmann, F.A.; Blech, M.; Hartl, J.; Garidel, P.; Hinderberger, D. Concentration Effects in the Interaction of Monoclonal Antibodies (mAbs) with their Immediate Environment Characterized by EPR Spectroscopy. Molecules 2019, 24, 2528. [Google Scholar] [CrossRef] [Green Version]
- Altenbach, C.; Kusnetzow, A.K.; Ernst, O.P.; Hofmann, K.P.; Hubbell, W.L. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. USA 2008, 105, 7439–7444. [Google Scholar] [CrossRef] [Green Version]
- Wingler, L.M.; Elgeti, M.; Hilger, D.; Latorraca, N.R.; Lerch, M.T.; Staus, D.P.; Dror, R.O.; Kobilka, B.K.; Hubbell, W.L.; Lefkowitz, R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Klare, J.P. Site-directed spin labeling EPR spectroscopy in protein research. Biol. Chem. 2013, 394, 1281–1300. [Google Scholar] [CrossRef]
- Columbus, L.; Hubbell, W.L. A new spin on protein dynamics. Trends Biochem. Sci. 2002, 27, 288–295. [Google Scholar] [CrossRef]
- Kusnetzow, A.K.; Altenbach, C.; Hubbell, W.L. Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry 2006, 45, 5538–5550. [Google Scholar] [CrossRef] [Green Version]
- Junk, M.J.; Spiess, H.W.; Hinderberger, D. Characterization of the Solution Structure of Human Serum Albumin Loaded with a Metal Porphyrin and Fatty Acids. Biophys. J. 2011, 100, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol.(Lausanne) 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, D.; Stichel, J.; Beck-Sickinger, A.G. Molecular recognition of the NPY hormone family by their receptors. Nutrition 2008, 24, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.; Berger, C.; Scheidt, H.A.; Berndt, S.; Bunge, A.; Beck-Sickinger, A.G.; Huster, D. A reconstitution protocol for the in vitro folded human G protein-coupled Y2 receptor into lipid environment. Biophys. Chem. 2010, 150, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.; Bender, B.J.; Kaiser, A.; Gulati, K.; Scheidt, H.A.; Hamm, H.E.; Meiler, J.; Beck-Sickinger, A.G.; Huster, D. Improved in Vitro Folding of the Y2 G Protein-Coupled Receptor into Bicelles. Front. Mol. Biosci. 2017, 4, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, K.; Kaiser, A.; Schmidt, P.; Splith, V.; Thomas, L.; Berndt, S.; Huster, D.; Beck-Sickinger, A.G. Oxidative in vitro folding of a cysteine deficient variant of the G protein-coupled neuropeptide Y receptor type 2 improves stability at high concentration. Biol. Chem. 2013, 394, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Muller, P.; Zellmann, T.; Scheidt, H.A.; Thomas, L.; Bosse, M.; Meier, R.; Meiler, J.; Huster, D.; Beck-Sickinger, A.G.; et al. Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y(2) Receptor Binding and Activation. Angew. Chem. Int. Ed. Engl. 2015, 54, 7446–7449. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kogler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524. [Google Scholar] [CrossRef]
- Wagner, S.; Klepsch, M.M.; Schlegel, S.; Appel, A.; Draheim, R.; Tarry, M.; Hogbom, M.; van Wijk, K.J.; Slotboom, D.J.; Persson, J.O.; et al. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. USA 2008, 105, 14371–14376. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Montag, C.; Berndt, S.; Huster, D. Optimization of Escherichia coli cultivation methods for high yield neuropeptide Y receptor type 2 production. Protein Expr. Purif. 2011, 76, 25–35. [Google Scholar] [CrossRef]
- Schrottke, S.; Kaiser, A.; Vortmeier, G.; Els-Heindl, S.; Worm, D.; Bosse, M.; Schmidt, P.; Scheidt, H.A.; Beck-Sickinger, A.G.; Huster, D. Expression, Functional Characterization, and Solid-State NMR Investigation of the G Protein-Coupled GHS Receptor in Bilayer Membranes. Sci. Rep. 2017, 7, 46128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, A.I.; Mileni, M.; Chien, E.Y.; Hanson, M.A.; Stevens, R.C. Microscale fluorescent thermal stability assay for membrane proteins. Structure 2008, 16, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Bosse, M.; Thomas, L.; Hassert, R.; Beck-Sickinger, A.G.; Huster, D.; Schmidt, P. Assessment of a fully active class A G protein-coupled receptor isolated from in vitro folding. Biochemistry 2011, 50, 9817–9825. [Google Scholar] [CrossRef]
- Kostenis, E.; Zeng, F.Y.; Wess, J. Functional characterization of a series of mutant G protein alphaq subunits displaying promiscuous receptor coupling properties. J. Biol. Chem. 1998, 273, 17886–17892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostenis, E. Potentiation of GPCR-signaling via membrane targeting of G protein alpha subunits. J. Recept. Signal. Transduct. Res. 2002, 22, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Hempel, C.; Wanka, L.; Schubert, M.; Hamm, H.E.; Beck-Sickinger, A.G. G Protein Preassembly Rescues Efficacy of W(6.48) Toggle Mutations in Neuropeptide Y(2) Receptor. Mol. Pharmacol. 2018, 93, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.; Lindner, D.; Montag, C.; Berndt, S.; Beck-Sickinger, A.G.; Rudolph, R.; Huster, D. Prokaryotic expression, in vitro folding, and molecular pharmacological characterization of the neuropeptide Y receptor type 2. Biotechnol. Prog. 2009, 25, 1732–1739. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Ge, M.T.; Rananavare, S.B.; Freed, J.H. ESR studies of stearic acid binding to bovine serum albumin. Biochim. Biophys. Acta 1990, 1036, 228–236. [Google Scholar]
- Freed, D.M.; Khan, A.K.; Horanyi, P.S.; Cafiso, D.S. Molecular origin of electron paramagnetic resonance line shapes on β-barrel membrane proteins: The local solvation environment modulates spin-label configuration. Biochemistry 2011, 50, 8792–8803. [Google Scholar] [CrossRef] [Green Version]
- Pavicevic, A.A.; Popovic-Bijelic, A.D.; Mojovic, M.D.; Susnjar, S.V.; Bacic, G.G. Binding of doxyl stearic spin labels to human serum albumin: An EPR study. J. Phys. Chem. B 2014, 118, 10898–10905. [Google Scholar] [CrossRef] [PubMed]
- Budil, D.E.; Earle, K.A.; Freed, D.M. Full determination of the rotational diffusion tensor by electron paramagnetic resonance at 250 GHz. J. Phys. Chem. 1993, 97, 1294–1303. [Google Scholar] [CrossRef]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis 2006—A comprehensive software package for analyzing pulsed ELDOR data. Appl. Mag. Res. 2006, 30, 473–498. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EC50 [nM] | pEC50 ± SEM | Emax ± SE [%] | |
|---|---|---|---|
| Y2R_eYFP_N1 (wt) | 0.3 | 9.59 ± 0.07 | 100 |
| Y2R_Δ6Cys | 0.6 | 9.25 ± 0.09 | 86 ± 2 |
| Y2R_Δ4Cys_C1513.53/C3167.44 | 0.4 | 9.45 ± 0.15 | 87 ± 3 |
| Y2R_Δ5Cys_C1513.53 | 0.8 | 9.09 ± 0.10 | 85 ± 3 |
| Y2R_Δ5Cys_C3167.44 | 0.4 | 9.45 ± 0.15 | 87 ± 3 |
| Y2R_Δ6Cys_A2025.21C | 2.2 | 8.66 ± 0.18 | 81 ± 5 |
| Y2R_Δ6Cys_R2626.29C | 1.1 | 8.94 ± 0.17 | 75 ± 4 |
| Y2R_Δ6Cys_L3007.28C | 2.0 | 8.70 ± 0.10 | 47 ± 2 |
| MTSL | IDSL | |||
|---|---|---|---|---|
| Conformation1 | Conformation2 | Conformation1 | Conformation2 | |
| A2025.21C (ECL) | ||||
| [Ax, Ay, Az] Aiso | [18.5, 18.3, 108.0] 48.3 | [18.5, 15.3, 95.7] 43.2 | [18.5, 18.3, 98.3] 45.0 | [18.5, 15.3, 88.7] 40.8 |
| τc | 5.5 | 4.7 | 9.9 | 2.9 |
| Pop% | 75.2 | 23.6 | 63.5 | 30.7 |
| L3007.26C (ECL) | ||||
| [Ax, Ay, Az] Aiso | [18.5, 18.3, 108.0] 48.3 | [19.5, 15.3, 95.7] 43.5 | [18.5, 14.3, 95.0] 42.6 | [15.5, 15.3, 93.7] 41.5 |
| τc | 5.1 | 4.4 | 35.9 | 4.7 |
| Pop% | 77.7 | 21.5 | 64.4 | 34.0 |
| R2626.29C (ICL) | ||||
| [Ax, Ay, Az] Aiso | [16.5, 18.3, 106.0] 46.9 | [16.5, 16.3, 95.7] 42.8 | [14.5, 15.3, 93.0] 40.9 | [15.3, 15.3, 90.7] 40.4 |
| τc | 15.6 | 6.6 | 36.3 | 4.3 |
| Pop% | 49.6 | 49.3 | 80.7 | 18.2 |
| C1513.53 (ICL) | ||||
| [Ax, Ay, Az] Aiso | [19.5, 15.3, 106.8] 47.2 | [19.5, 15.3, 95.7] 43.5 | [19.5, 14.3, 95.8] 43.2 | [18.5, 18.3, 90.7] 42.5 |
| τc | 6.0 | 2.6 | 15.3 | 4.9 |
| Pop% | 85.3 | 14.2 | 87.2 | 11.5 |
| C3167.44 (TM) | ||||
| [Ax, Ay, Az] Aiso | [19.5, 14.3, 111.0] 48.3 | [19.5, 15.3, 95.7] 43.5 | [19.5, 14.3, 95.0] 42.9 | [18.5, 15.3, 95.7] 43.2 |
| τc | 5.7 | 1.1 | 15.3 | 4.7 |
| Pop% | 93.7 | 6.2 | 82.2 | 16.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laugwitz, J.M.; Haeri, H.H.; Kaiser, A.; Krug, U.; Hinderberger, D.; Beck-Sickinger, A.G.; Schmidt, P. Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements. Molecules 2020, 25, 4143. https://doi.org/10.3390/molecules25184143
Laugwitz JM, Haeri HH, Kaiser A, Krug U, Hinderberger D, Beck-Sickinger AG, Schmidt P. Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements. Molecules. 2020; 25(18):4143. https://doi.org/10.3390/molecules25184143
Chicago/Turabian StyleLaugwitz, Jeannette M., Haleh H. Haeri, Anette Kaiser, Ulrike Krug, Dariush Hinderberger, Annette G. Beck-Sickinger, and Peter Schmidt. 2020. "Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements" Molecules 25, no. 18: 4143. https://doi.org/10.3390/molecules25184143
APA StyleLaugwitz, J. M., Haeri, H. H., Kaiser, A., Krug, U., Hinderberger, D., Beck-Sickinger, A. G., & Schmidt, P. (2020). Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements. Molecules, 25(18), 4143. https://doi.org/10.3390/molecules25184143