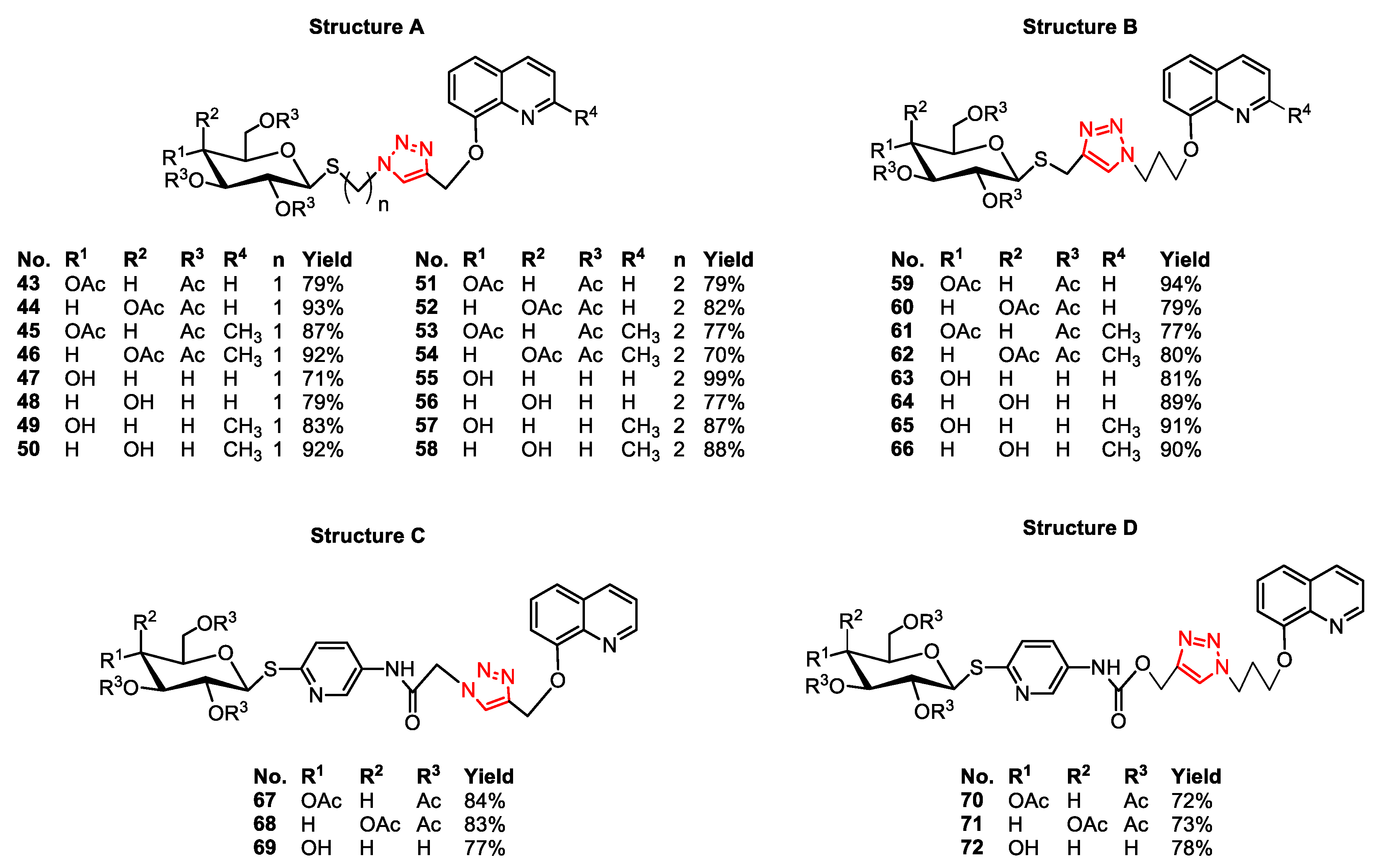
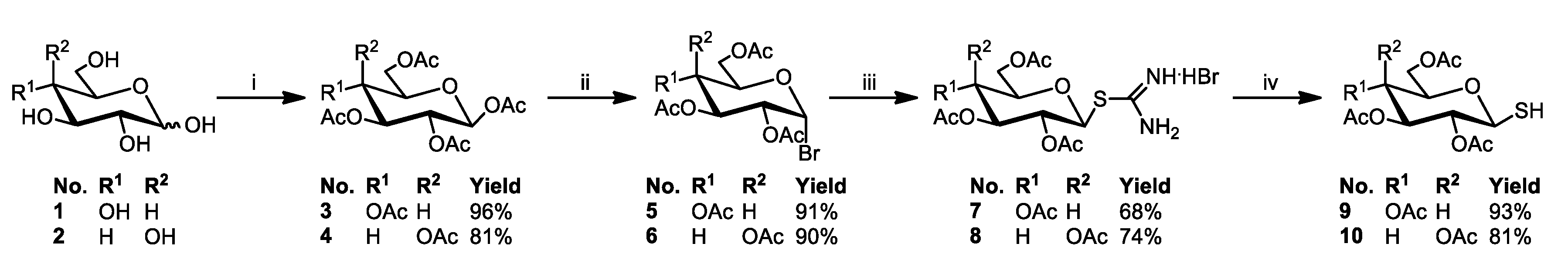
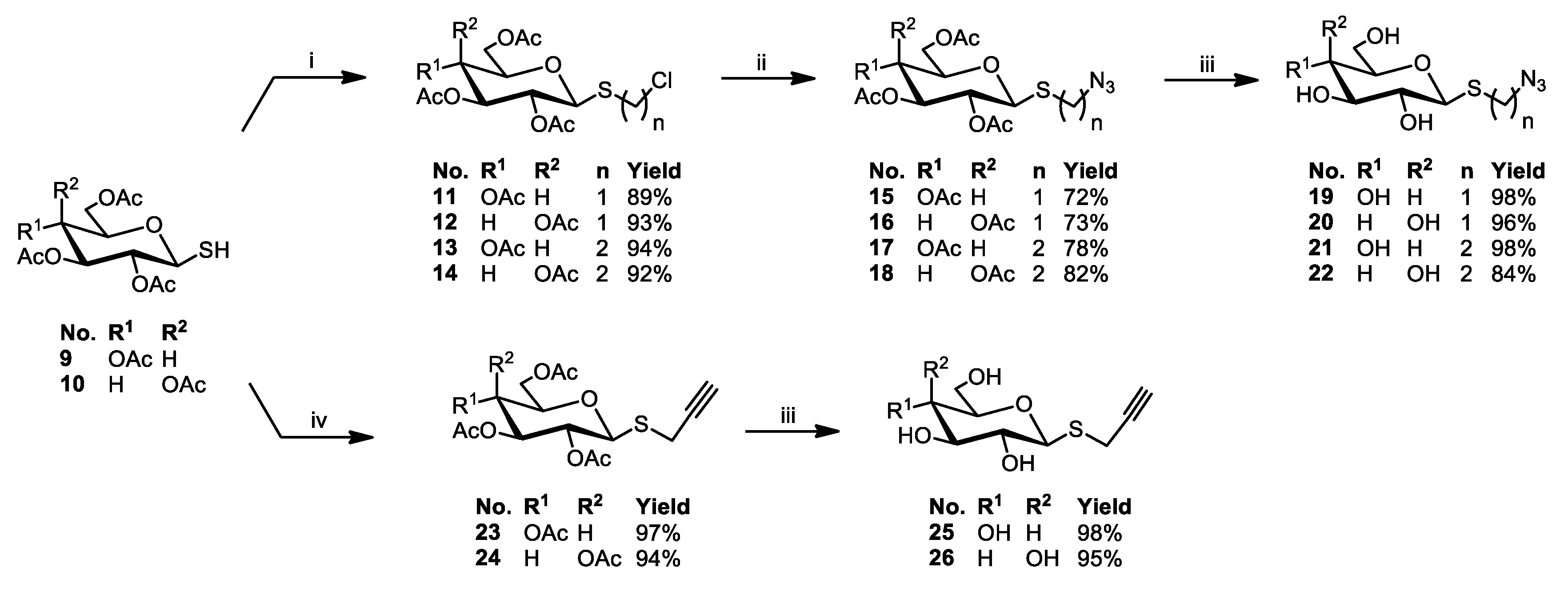
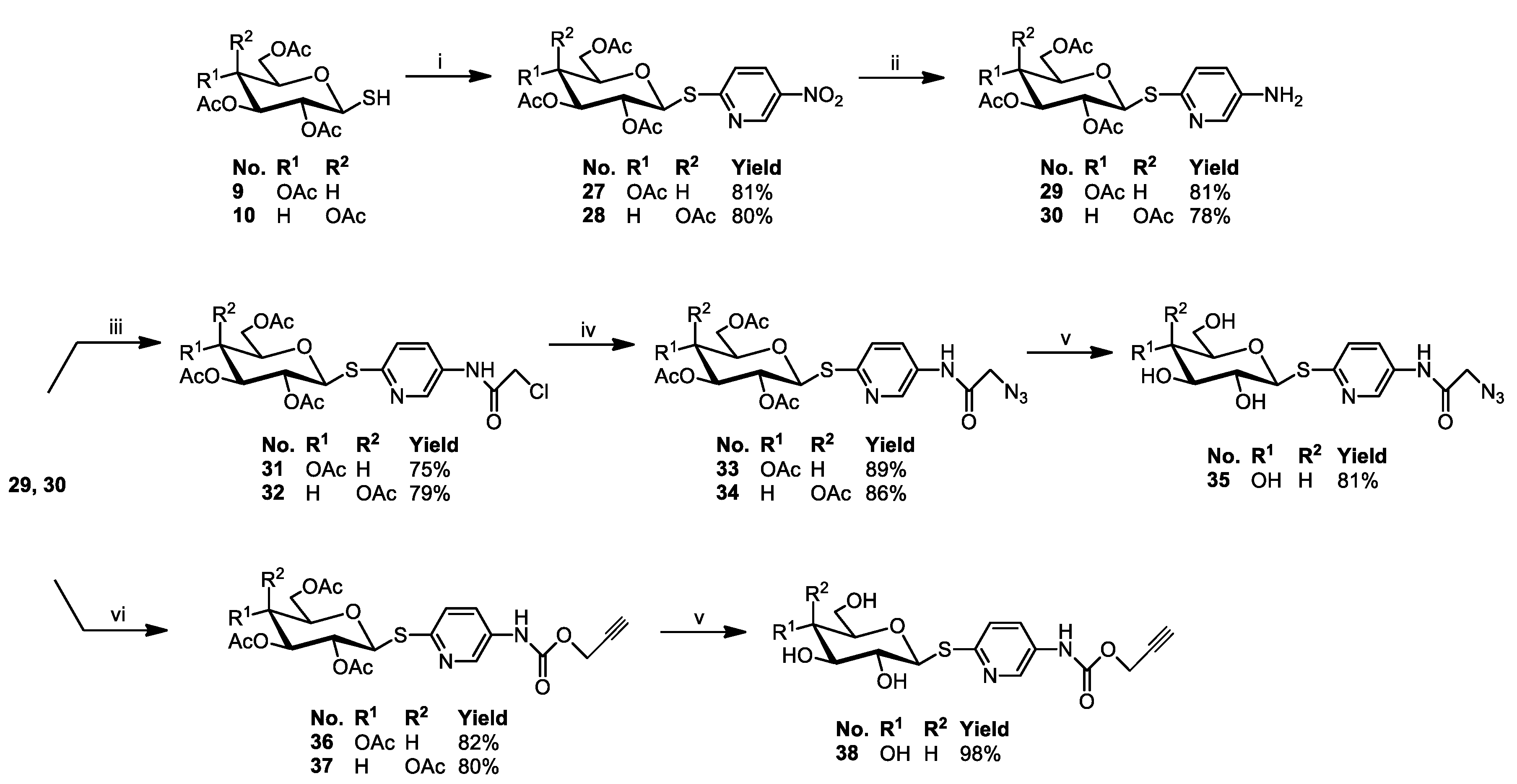
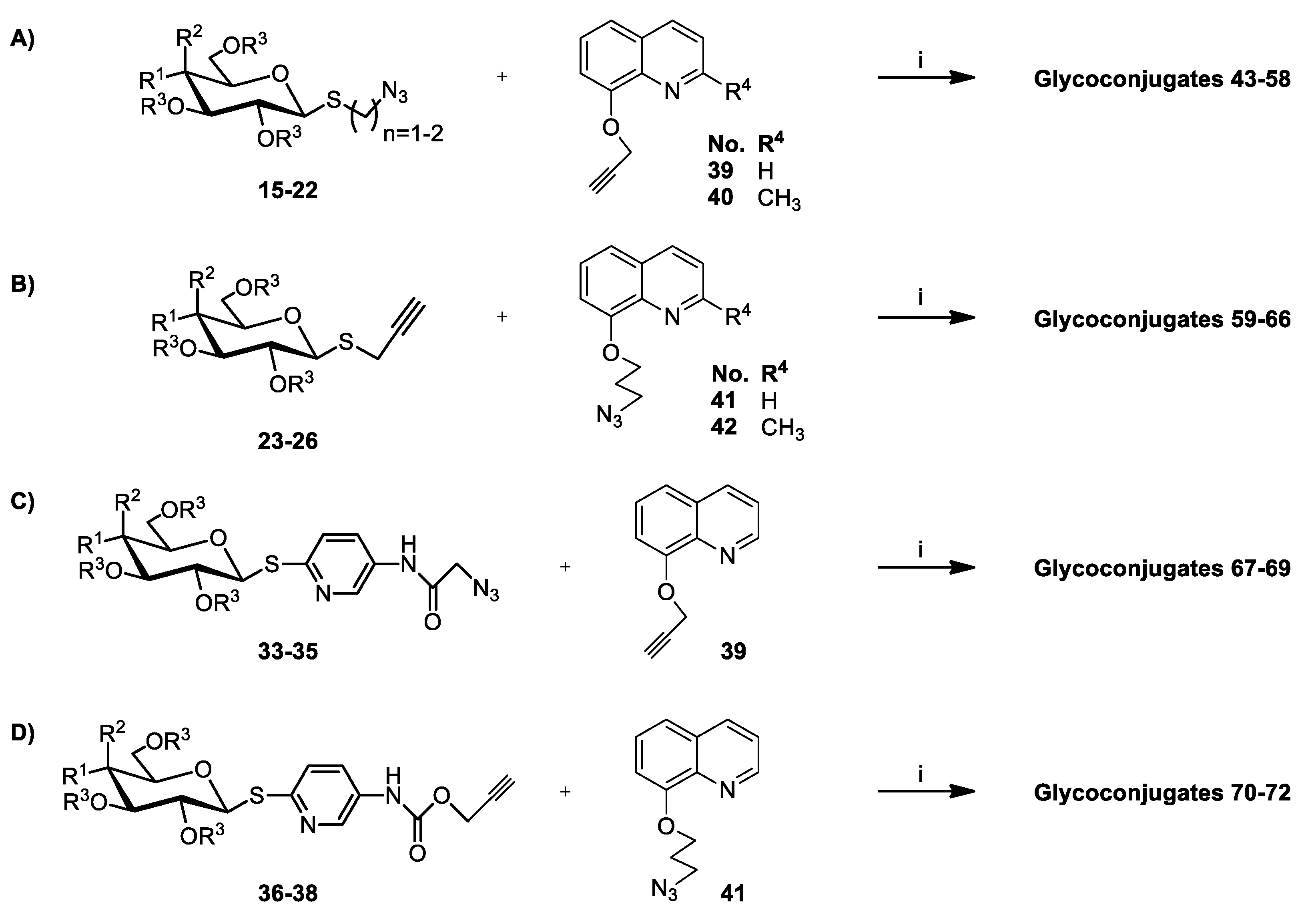
3.2.5. General Procedure for the Synthesis of Glycoconjugates 43–72
The appropriate sugar derivatives 15–26, 33–38 (1 eq.) and 8-hydroxyquinoline derivatives 39–41 (1 eq.) were dissolved in a dry solvent system: THF (2 mL) and i-PrOH (2 mL). The catalyst systems were prepared: sodium ascorbate (0.4 eq.) dissolved in H2O (1 mL) and CuSO4·5H2O (0.2 eq.) dissolved in H2O (1 mL), mixed, and immediately added to the reaction mixture. The reaction mixture was stirred for 24 h at room temperature. Then, the solvents were evaporated in vacuo, and the crude products were purified by column chromatography (dry loading; toluene:AcOEt, 2:1 and CHCl3:MeOH, 100:1 for fully protected glycoconjugates or CHCl3:MeOH, gradient: 50:1 to 2:1 for glycoconjugates with unprotected sugar part) to give products 43–72.
Glycoconjugate43: Starting from sugar derivative15and 8-HQ derivative39, the product was obtained as a beige solid (79% yield), m.p.: 191–196 °C; [α]23D = −70.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.94, 1.99, 2.02, 2.05 (4s, 12H, CH3CO), 3.61 (ddd, 1H, J = 2.2 Hz, J = 4.8 Hz, J = 10.0 Hz, H-5Glu), 4.05 (dd, 1H, J = 2.2 Hz, J = 12.5 Hz, H-6aGlu), 4.17 (dd, 1H, J = 4.8 Hz, J = 12.5 Hz, H-6bGlu), 4.55 (d, 1H, J = 10.1 Hz, H-1Glu), 5.02 (dd~t, 1H, J = 9.2 Hz, J = 10.0 Hz, H-4Glu), 5.06 (dd~t, 1H, J = 9.4 Hz, J = 10.1 Hz, H-2Glu), 5.15 (dd~t, 1H, J = 9.2 Hz, J = 9.4 Hz, H-3Glu), 5.29 i 5.71 (qAB, 2H, J = 14.5 Hz, CH2N3), 5.58 (s, 2H, CH2O), 7.29 (dd, 1H, J = 1.9 Hz, J = 6.9 Hz, H-7Quin), 7.40–7.49 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.94 (s, 1H, H-5Triaz), 8.14 (dd, 1H, J = 1.6 Hz, J = 8.3 Hz, H-4Quin), 8.93 (dd, 1H, J = 1.6 Hz, 4.1 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ 20.51, 20.55, 20.57, 20.74 (CH3CO), 48.05 (CH2N3), 61.58, 62.85 (C-6Glu, CH2O), 67.91, 69.77, 73.56, 76.20 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 81.48 (C-1Glu), 109.93 (C-7Quin), 120.44 (C-5Quin), 121.71 (C-3Quin), 123.20 (C-5Triaz), 126.68 (C-6Quin), 129.52 (C-4aQuin), 136.00 (C-4Quin), 140.32 (C-8aQuin), 145.12 (C-4Triaz), 149.42 (C-2Quin), 155.63 (C-8Quin), 169.35, 169.40, 169.98, 170.54 (CH3CO); HRMS (ESI-TOF): calcd for C27H31N4O10S ([M + H]+): m/z 603.1761; found: m/z 603.1766.
Glycoconjugate44: Starting from sugar derivative16and 8-HQ derivative39, the product was obtained as a beige solid (93% yield), m.p.: 92–96 °C; [α]23D = −55.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.95, 1.97, 2.03, 2.15 (4s, 12H, CH3CO), 3.67 (ddd, 1H, J = 0.8 Hz, J = 6.0 Hz, J = 6.9 Hz, H-5Gal), 3.93 (dd, 1H, J = 6.0 Hz, J = 11.4 Hz, H-6aGal), 3.98 (dd, 1H, J = 6.9 Hz, J = 11.4 Hz, H-6bGal), 4.46 (d, 1H, J = 10.0 Hz, H-1Gal), 4.95 (dd, 1H, J = 3.4 Hz, J = 10.0 Hz, H-3Gal), 5.21 (dd~t, 1H, J = 10.0 Hz, J = 10.0 Hz, H-2Gal), 5.31 and 5.71 (qAB, 2H, J = 14.6 Hz, CH2N3), 5.33 (dd, 1H, J = 0.8 Hz, J = 3.4 Hz, H-4Gal), 5.60 (s, 2H, CH2O), 7.28 (dd, 1H, J = 2.7 Hz, J = 6.4 Hz, H-7Quin), 7.41–7.48 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.94 (s, 1H, H-5Triaz), 8.15 (dd, 1H, J = 1.6 Hz, J = 8.3 Hz, H-4Quin), 8.93 (dd, 1H, J = 1.6 Hz, 4.1 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ20.52, 20.60, 20.65, 20.69 (CH3CO), 47.93 (CH2N3), 61.36, 62.85 (C-6Gal, CH2O), 66.98, 67.13, 71.54, 74.92 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 81.81 (C-1Gal), 109.96 (C-7Quin), 120.40 (C-5Quin), 121.74 (C-3Quin), 123.25 (C-5Triaz), 126.69 (C-6Quin), 129.53 (C-4aQuin), 135.98 (C-4Quin), 140.34 (C-8aQuin), 144.99 (C-4Triaz), 149.45 (C-2Quin), 153.71 (C-8Quin), 169.60, 169.81, 170.12, 170.26 (CH3CO); HRMS (ESI-TOF): calcd for C27H31N4O10S ([M + H]+): m/z 603.1761; found: m/z 603.1760.
Glycoconjugate45: Starting from sugar derivative15and 8-HQ derivative40, the product was obtained as a white solid (87% yield), m.p.: 159–163 °C; [α]23D = −84.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.94, 1.99, 2.02, 2.07 (4s, 12H, CH3CO), 2.78 (s, 3H, CH3), 3.55 (ddd, 1H, J = 1.9 Hz, J = 4.8 Hz, J = 10.1 Hz, H-5Glu), 4.01 (dd, 1H, J = 1.9 Hz, J = 12.5 Hz, H-6aGlu), 4.15 (dd, 1H, J = 4.8 Hz, J = 12.5 Hz, H-6bGlu), 4.52 (d, 1H, J = 10.1 Hz, H-1Glu), 5.01 (dd~t, 1H, J = 9.3 Hz, J = 10.1 Hz, H-4Glu), 5.04 (dd~t, 1H, J = 9.3 Hz, J = 10.1 Hz, H-2Glu), 5.14 (dd~t, 1H, J = 9.3 Hz, J = 9.3 Hz, H-3Glu), 5.28 i 5.71 (qAB, 2H, J = 14.6 Hz, CH2N3), 5.59 i 5.62 (qAB, 2H, J = 13.4 Hz, CH2O), 7.22 (dd, 1H, J = 1.4 Hz, J = 7.2 Hz, H-7Quin), 7.31 (d, 1H, J = 8.4 Hz, H-3Quin), 7.34–7.40 (m, 2H, H-5Quin, H-6Quin), 7.92 (s, 1H, H-5Triaz), 8.01 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.51, 20.55, 20,57, 20.74 (CH3CO), 25.74 (CH3), 47.97 (CH2N), 61.57, 63.21 (C-6Glu, CH2O), 67.90, 69.79, 73.55, 76.17 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 81.33 (C-1Glu), 110.48 (C-7Quin), 120.41 (C-5Quin), 122.62 (C-3Quin), 123.04 (C-5Triaz), 125.61 (C-6Quin), 127.76 (C-4aQuin), 136.17 (C-4Quin), 139.89 (C-8aQuin), 145.46 (C-4Triaz), 153.23 (C-2Quin), 158.29 (C-8Quin), 169.33, 169.40, 169.96, 170.51 (CH3CO); HRMS (ESI-TOF): calcd for C28H33N4O10S ([M + H]+): m/z 617.1917; found: m/z 617.1915.
Glycoconjugate46: Starting from sugar derivative16and 8-HQ derivative40, the product was obtained as a beige solid (92% yield), m.p.: 65–68 °C; [α]23D = −34.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.95, 1.97, 2.06, 2.14 (4s, 12H, CH3CO), 2.78 (s, 3H, CH3), 3.49 (ddd, 1H, J = 0.8 Hz, J = 5.7 Hz, J = 7.1 Hz, H-5Gal), 3.83 (dd, 1H, J = 5.7 Hz, J = 11.5 Hz, H-6aGal), 3.93 (dd, 1H, J = 7.1 Hz, J = 11.5 Hz, H-6bGal), 4.38 (d, 1H, J = 10.0 Hz, H-1Gal), 4.93 (dd, 1H, J = 3.4 Hz, J = 10.0 Hz, H-3Gal), 5.19 (dd~t, 1H, J = 10.0 Hz, J = 10.0 Hz, H-2Gal), 5.28 (dd, 1H, J = 0.8 Hz, J = 3.4 Hz, H-4Gal), 5.30 and 5.70 (qAB, 2H, J = 14.5 Hz, CH2N3), 5.61 (dd, 2H, J = 2.5 Hz, J = 16.5 Hz, CH2O), 7.20 (dd, 1H, J = 1.4 Hz, J = 7.4 Hz, H-7Quin), 7.31 (d, 1H, J = 8.4 Hz, H-3Quin), 7.34–7.43 (m, 2H, H-5Quin, H-6Quin), 7.91 (s, 1H, H-5Triaz), 8.02 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.52, 20.60, 20.63, 20.69 (CH3CO), 25.71 (CH3) 47.77 (CH2N3), 61.49, 63.13 (C-6Gal, CH2O), 67.03, 67.16, 71.51, 74.85 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 81.51 (C-1Gal), 110.47 (C-7Quin), 120.38 (C-5Quin), 122.70 (C-3Quin), 123.20 (C-5Triaz), 125.62 (C-6Quin), 127.77 (C-4aQuin), 136.15 (C-4Quin), 139.90 (C-8aQuin), 145.27 (C-4Triaz), 153.14 (C-2Quin), 158.37 (C-8Quin), 169.60, 169.80, 170.10, 170.24 (CH3CO); HRMS (ESI-TOF): calcd for C28H33N4O10S ([M + H]+): m/z 617.1917; found: m/z 617.1917.
Glycoconjugate47: Starting from sugar derivative19and 8-HQ derivative39, the product was obtained as a white solid (71% yield), m.p.: 123–126 °C; [α]23D = −127.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 3.02–3.10 (m, 2H, H-2Glu, H-4Glu), 3.16 (m, 1H, H-5Glu), 3.26 (m, 1H, H-3Glu), 3.48 (m, 1H, H-6aGlu), 3.78 (m, 1H, H-6bGlu), 4.47 (d, 1H, J = 9.7 Hz, H-1Glu), 4.96 (t, 1H, J = 5.4 Hz, 6-OH), 5.02 (d, 1H, J = 5.5 Hz, OH), 5.10 (d, 1H, J = 4.9 Hz, OH), 5.29 (d, 1H, J = 6.2 Hz, OH), 5.36 (s, 2H, CH2O), 5.65 and 5.82 (qAB, 2H, J = 14.5 Hz, CH2N3), 7.42 (dd, 1H, J = 3.4 Hz, J = 5.4 Hz, H-7Quin), 7.51–7.58 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 8.34 (dd, 1H, J = 1.4 Hz, J = 8.3 Hz, H-4Quin), 8.56 (s, 1H, H-5Triaz), 8.83 (d, 1H, J = 2.4 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 47.81 (CH2N3), 61.39, 61.97 (C-6Glu, CH2O), 70.16, 73.25, 78.05, 81.25 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 83.88 (C-1Glu), 110.01 (C-7Quin), 120.04 (C-5Quin), 121.88 (C-3Quin), 125.28 (C-5Triaz), 126.79 (C-6Quin), 129.08 (C-4aQuin), 135.97 (C-4Quin), 139.55 (C-8aQuin), 142.95 (C-4Triaz), 148.96 (C-2Quin), 153.74 (C-8Quin); HRMS (ESI-TOF): calcd for C19H23N4O6S ([M + H]+): m/z 435.1338; found: m/z 435.1339.
Glycoconjugate48: Starting from sugar derivative20and 8-HQ derivative39, the product was obtained as a beige solid (79% yield), m.p.: 142–145 °C; [α]23D = −58.7 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 3.29 (m, 1H, H-3Gal), 3.39 (m, 1H, H-5Gal), 3.45 (m, 1H, H-2Gal), 3.53 (m, 1H, H-6aGal), 3.60 (m, 1H, H-6bGal), 3.69 (m, 1H, H-4Gal), 4.40 (d, 1H, J = 9.6 Hz, H-1Gal), 4.52 (d, 1H, J = 4.5 Hz, OH), 4.84 (d, 1H, J = 5.2 Hz, OH), 4.91 (t, 1H, J = 5.2 Hz, 6-OH), 5.12 (d, 1H, J = 6.1 Hz, OH), 5.36 (s, 2H, CH2O), 5.63 and 5.79 (qAB, 2H, J = 14.5 Hz, CH2N3), 7.41 (dd, 1H, J = 3.6, J = 5.4 Hz, H-7Quin), 7.50–7.58 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 8.33 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4Quin), 8.52 (s, 1H, H-5Triaz), 8.83 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 47.88 (CH2N), 60.89, 61.98 (C-6Gal, CH2O), 68.61, 70.01, 74.55, 79.68 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.39 (C-1Gal), 110.03 (C-7Quin), 120.04 (C-5Quin), 121.88 (C-3Quin), 125.20 (C-5Triaz), 126.79 (C-6Quin), 129.08 (C-4aQuin), 135.95 (C-4Quin), 139.57 (C-8aQuin), 142.91 (C-4Triaz), 148.96 (C-2Quin), 153.72 (C-8Quin); HRMS (ESI-TOF): calcd for C19H23N4O6S ([M + H]+): m/z 435.1338; found: m/z 435.1333.
Glycoconjugate49: Starting from sugar derivative19and 8-HQ derivative40, the product was obtained as a white solid (83% yield), m.p.: 140–144 °C; [α]23D = −64.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 2.64 (s, 3H, CH3), 3.02–3.10 (m, 2H, H-2Glu, H-4Glu), 3.15 (m, 1H, H-5Glu), 3.24 (m, 1H, H-3Glu), 3.45 (m, 1H, H-6aGlu), 3.75 (m, 1H, H-6bGlu), 4.46 (d, 1H, J = 9.7 Hz, H-1Glu), 4.77 (t, 1H, J = 5.5 Hz, 6-OH), 5.01 (d, 1H, J = 5.4 Hz, OH), 5.10 (d, 1H, J = 4.8 Hz, OH), 5.28 (d, 1H, J = 6.2 Hz, OH), 5.35 (s, 2H, CH2O), 5.64 and 5.81 (qAB, 2H, J = 14.5 Hz, CH2N3), 7.37 (dd, 1H, J = 1.4 Hz, J = 7.6, H-7Quin), 7.42 (d, 1H, J = 8.3 Hz, H-3Quin), 7.44 (dd~t, 1H, J = 7.3 Hz, J = 7.6 Hz, H-6Quin), 7.47 (dd, 1H, J = 1.2 Hz, J = 8.1 Hz, H-5Quin), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin), 8.49 (s, 1H, H-5Triaz); 13C NMR (100 MHz, DMSO): δ 24.88 (CH3), 47.69 (CH2N3), 61.31, 61.65 (C-6Glu, CH2O), 70.10, 73.24, 78.04, 81.27 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 83.74 (C-1Glu), 110.22 (C-7Quin), 119.85 (C-5Quin), 122.49 (C-3Quin), 125.40, 125.66 (C-5Triaz, C-6Quin), 127.35 (C-4aQuin), 136.03 (C-4Quin), 139.14 (C-8aQuin), 142.91 (C-4Triaz), 153.29 (C-2Quin), 157.33 (C-8Quin); HRMS (ESI-TOF): calcd for C20H25N4O6S ([M + H]+): m/z 449.1495; found: m/z 449.1494.
Glycoconjugate50: Starting from sugar derivative20and 8-HQ derivative40, the product was obtained as a beige solid (92% yield), m.p.: 134–138 °C; [α]23D = −90.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 2.64 (s, 3H, CH3), 3.27 (m, 1H, H-3Gal), 3.34–3.44 (m, 2H, H-2Gal, H-5Gal), 3.49 (m, 1H, H-6aGal), 3.55 (m, 1H, H-6bGal), 3.67 (m, 1H, H-4Gal), 4.37 (d, 1H, J = 9.6 Hz, H-1Gal), 4.48 (d, 1H, J = 4.6 Hz, OH), 4.74 (t, 1H, J = 5.4 Hz, 6-OH), 4.84 (d, 1H, J = 4.9 Hz, OH), 5.11 (d, 1H, J = 6.1 Hz, OH), 5.36 (s, 2H, CH2O), 5.62 and 5.78 (qAB, 2H, J = 14.5 Hz, CH2N3), 7.36 (dd, 1H, J = 1.7 Hz, J = 7.4 Hz, H-7Quin), 7.39–7.44 (m, 2H, H-3Quin, H-6Quin), 7.47 (dd, 1H, J = 1.6 Hz, J = 8.1 Hz, H-5Quin), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin), 8.44 (s, 1H, H-5Triaz); 13C NMR (100 MHz, DMSO): δ 24.89 (CH3), 47.73 (CH2N3), 60.77, 61.65 (C-6Gal, CH2O), 68.53, 70.01, 74.54, 79.66 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.21 (C-1Gal), 110.26 (C-7Quin), 119.84 (C-5Quin), 122.49 (C-3Quin), 125.32, 125.66 (C-5Triaz, C-6Quin), 127.35 (C-4aQuin), 136.01 (C-4Quin), 139.17 (C-8aQuin), 142.87 (C-4Triaz), 153.25 (C-2Quin), 157.33 (C-8Quin); HRMS (ESI-TOF): calcd for C20H25N4O6S ([M + H]+): m/z 449.1495; found: m/z 449.1496.
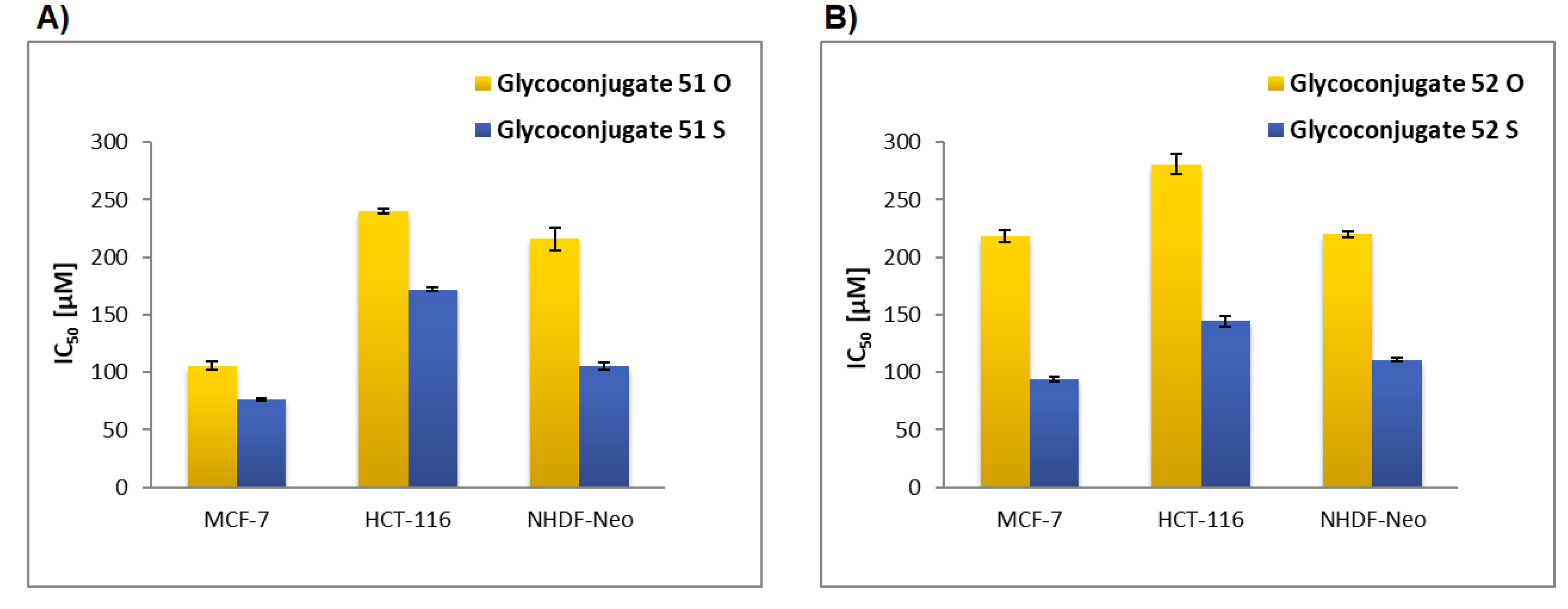
Glycoconjugate51: Starting from sugar derivative17and 8-HQ derivative39, the product was obtained as a brown solid (79% yield), m.p.: 150–152 °C; [α]23D = −31.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.00, 2.01, 2.03, 2.03 (4s, 12H, CH3CO), 3.05 (m, 1H, CHS), 3.25 (m, 1H, CHS), 3.70 (ddd, 1H, J = 2.8 Hz, J = 4.6 Hz, J = 10.1 Hz, H-5Glu), 4.13 (dd, 1H, J = 2.8 Hz, J = 12.5 Hz, H-6aGlu), 4.19 (dd, 1H, J = 4.6 Hz, J = 12.5 Hz, H-6bGlu), 4.47 (d, 1H, J = 10.0 Hz, H-1Glu), 4.50–4.67 (m, 2H, CH2N3), 5.02 (dd~t, 1H, J = 9.4 Hz, J = 10.1 Hz, H-4Glu), 5.04 (dd~t, 1H, J = 9.4 Hz, J = 10.0 Hz, H-2Glu), 5.21 (dd~t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3Glu), 5.58 (s, 2H, CH2O), 7.32 (dd, 1H, J = 1.3 Hz, J = 7.2 Hz, H-7Quin), 7.38–7.49 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.85 (s, 1H, H-5Triaz), 8.13 (dd, 1H, J = 1.4 Hz, J = 8.3 Hz, H-4Quin), 8.93 (d, 1H, J = 2.7 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ 20.56, 20.57, 20.63, 20.66 (CH3CO), 30.28 (CH2S) 50.61 (CH2N3), 61.85, 62.97 (C-6Glu, CH2O), 68.11, 69.50, 73.56, 76.25 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 83.73 (C-1Glu), 110.07 (C-7Quin), 120.27 (C-5Quin), 121.65 (C-3Quin), 124.05 (C-5Triaz), 126.74 (C-6Quin), 129.51 (C-4aQuin), 135.98 (C-4Quin), 140.37 (C-8aQuin), 144.03 (C-4Triaz), 149.37 (C-2Quin), 153.87 (C-8Quin), 169.35, 169.38, 170.04, 170.49 (CH3CO); HRMS (ESI-TOF): calcd for C28H33N4O10S ([M + H]+): m/z 617.1917; found: m/z 617.1918.
Glycoconjugate52: Starting from sugar derivative18and 8-HQ derivative39, the product was obtained as a yellow solid (82% yield), m.p.: 90–95 °C; [α]23D = −9.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 1.99, 2.04, 2.15 (4s, 12H, CH3CO), 3.06 (m, 1H, CHS), 3.28 (m, 1H, CHS), 3.92 (ddd, 1H, J = 0.8 Hz, J = 6.0 Hz, J = 6.9 Hz, H-5Gal), 4.08 (dd, 1H, J = 6.0 Hz, J = 11.7 Hz, H-6aGal), 4.12 (dd, 1H, J = 6.9 Hz, J = 11.7 Hz, H-6bGal), 4.46 (d, 1H, J = 9.9 Hz, H-1Gal), 4.53–4.69 (m, 2H, CH2N3), 5.03 (dd, 1H, J = 3.3 Hz, J = 10.0 Hz, H-3Gal), 5.24 (dd~t, 1H, J = 9.9 Hz, J = 10.0 Hz, H-2Gal), 5.43 (dd, 1H, J = 0.8 Hz, J = 3.3 Hz, H-4Gal), 5.58 (s, 2H, CH2O), 7.34 (d, 1H, J = 7.1 Hz, H-7Quin), 7.38–7.49 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.85 (s, 1H, H-5Triaz), 8.13 (d, 1H, J = 8.0 Hz, H-4Quin), 8.94 (d, 1H, J = 1.9 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ 20.56, 20.63, 20.68, 20.74 (CH3CO), 30.29 (CH2S) 50.66 (CH2N), 61.61, 62.93 (C-6Gal, CH2O), 66.76, 67.24, 71.64, 74.90 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.17 (C-1Gal), 109.99 (C-7Quin), 120.27 (C-5Quin), 121.66 (C-3Quin), 123.86 (C-5Triaz), 126.74 (C-6Quin), 129.51 (C-4aQuin), 135.96 (C-4Quin), 140.36 (C-8aQuin), 144.09 (C-4Triaz), 149.37 (C-2Quin), 153.89 (C-8Quin), 169.61, 169.94, 170.13, 170.33 (CH3CO); HRMS (ESI-TOF): calcd for C28H33N4O10S ([M + H]+): m/z 617.1917; found: m/z 617.1920.
Glycoconjugate53: Starting from sugar derivative17and 8-HQ derivative40, the product was obtained as a beige solid (77% yield), m.p.: 139–142 °C; [α]23D = −29.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.00, 2.01, 2.02, 2.03 (4s, 12H, CH3CO), 2.79 (s, 3H, CH3), 3.05 (m, 1H, CHS), 3.25 (m, 1H, CHS), 3.70 (ddd, 1H, J = 2.9 Hz, J = 4.5 Hz, J = 10.1 Hz, H-5Glu), 4.12 (dd, 1H, J = 2.9 Hz, J = 12.5 Hz, H-6aGlu), 4.18 (dd, 1H, J = 4.5 Hz, J = 12.5 Hz, H-6bGlu), 4.47 (d, 1H, J = 10.0 Hz, H-1Glu), 4.50–4.67 (m, 2H, CH2N3), 5.01 (dd~t, 1H, J = 9.4 Hz, J = 10.1 Hz, H-4Glu), 5.04 (dd~t, 1H, J = 9.4 Hz, J = 10.0 Hz, H-2Glu), 5.20 (dd~t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3Glu), 5.59 (s, 2H, CH2O), 7.26 (dd, 1H, J = 3.3 Hz, J = 5.6 Hz, H-7Quin), 7.31 (d, 1H, J = 8.4 Hz, H-3Quin), 7.33–7.39 (m, 2H, H5Quin, H-6Quin), 7.83 (s, 1H, H-5Triaz), 8.01 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.56, 20.57, 20.63, 20.66 (CH3CO), 25.73 (CH3), 30.33 (CH2S) 50.60 (CH2N3), 61.86, 63.33 (C-6Glu, CH2O), 68.12, 69.50, 73.57, 76.24 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 83.74 (C-1Glu), 110.65 (C-7Quin), 120.26 (C-5Quin), 122.59 (C-3Quin), 123.91 (C-5Triaz), 125.70 (C-6Quin), 127.76 (C-4aQuin), 136.17 (C-4Quin), 139.93 (C-8aQuin), 144.37 (C-4Triaz), 153.37 (C-2Quin), 158.19 (C-8Quin), 169.36, 169.40, 170.04, 170.50 (CH3CO); HRMS (ESI-TOF): calcd for C29H35N4O10S ([M + H]+): m/z 631.2074; found: m/z 631.2075.
Glycoconjugate54: Starting from sugar derivative18and 8-HQ derivative40, the product was obtained as a white solid (70% yield), m.p.: 56–61 °C; [α]23D = −9.3 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 1.99, 2.04, 2.15 (4s, 12H, CH3CO), 2.79 (s, 3H, CH3), 3.06 (m, 1H, CHS), 3.28 (m, 1H, CHS), 3.90 (ddd, 1H, J = 0.7 Hz, J = 6.0 Hz, J = 7.0 Hz, H-5Gal), 4.07 (dd, 1H, J = 6.0 Hz, J = 11.9 Hz, H-6aGal), 4.11 (dd, 1H, J = 7.0 Hz, J = 11.9 Hz, H-6bGal), 4.45 (d, 1H, J = 9.9 Hz, H-1Gal), 4.53–4.69 (m, 2H, CH2N3), 5.02 (dd, 1H, J = 3.3 Hz, J = 10.0 Hz, H-3Gal), 5.24 (dd~t, 1H, J = 9.9 Hz, J = 10.0 Hz, H-2Gal), 5.42 (dd, 1H, J = 0.7 Hz, J = 3.3 Hz, H-4Gal), 5.60 (s, 2H, CH2O), 7.28 (m, 1H, H-7Quin), 7.31 (d, 1H, J = 8.4 Hz, H-3Quin), 7.33–7.39 (m, 2H, H-5Quin, H-6Quin), 7.83 (s, 1H, H-5Triaz), 8.01 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.56, 20.63, 20.67, 20.74 (CH3CO), 25.79 (CH3), 30.30 (CH2S) 50.65 (CH2N), 61.62, 63.33 (C-6Gal, CH2O), 66.75, 67.24, 71.64, 74.89 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.15 (C-1Gal), 110.59 (C-7Quin), 120.25 (C-5Quin), 122.58 (C-3Quin), 123.69 (C-5Triaz), 125.70 (C-6Quin), 127.75 (C-4aQuin), 136.17 (C-4Quin), 139.90 (C-8aQuin), 144.44 (C-4Triaz), 153.36 (C-2Quin), 158.20 (C-8Quin), 169.61, 169.93, 170.12, 170.33 (CH3CO); HRMS (ESI-TOF): calcd for C29H35N4O10S ([M + H]+): m/z 631.2074; found: m/z 631.2070.
Glycoconjugate55: Starting from sugar derivative21and 8-HQ derivative39, the product was obtained as a brown solid (99% yield), m.p.: 65–69 °C; [α]23D = −4.0 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 2.97–3.24 (m, 6H, H-2Glu, H-3Glu, H-4Glu H-5Glu, CH2S), 3.45 (m, 1H, 6aGlu), 3.72 (m, 1H, 6bGlu), 4.37 (d, 1H, J = 9.6 Hz, H-1Glu), 4.57–4.73 (m, 3H, CH2N3, 6-OH), 4.95 (d, 1H, J = 5.3 Hz, OH), 5.03 (d, 1H, J = 4.8 Hz, OH), 5.21 (d, 1H, J = 5.8 Hz, OH), 5.35 (s, 2H, CH2O), 7.41 (dd, 1H, J = 3.8 Hz, J = 5.3 Hz, H-7Quin), 7.50–7.57 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 8.32 (dd, 1H, J = 1.8 Hz, J = 8.3 Hz, H-4Quin), 8.34 (s, 1H, H-5Triaz), 8.83 (dd, 1H, J = 1.8 Hz, J = 4.1 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 29.78 (CH2S), 49.91 (CH2N3), 61.28, 61.91 (C-6Glu, CH2O), 70.06, 72.95, 78.10, 81.00 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 85.36 (C-1Glu), 110.06 (C-7Quin), 120.01 (C-5Quin), 121.84 (C-3Quin), 125.39 (C-5Triaz), 126.75 (C-6Quin), 129.06 (C-4aQuin), 135.81 (C-4Quin), 139.70 (C-8aQuin), 142.21 (C-4Triaz), 148.95 (C-2Quin), 153.86 (C-8Quin); HRMS (ESI-TOF): calcd for C20H25N4O6S ([M + H]+): m/z 449.1495; found: m/z 449.1492.
Glycoconjugate56: Starting from sugar derivative22and 8-HQ derivative39, the product was obtained as a beige solid (77% yield), m.p.: 125–128 °C; [α]23D = 6.2 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 3.06 (m, 1H, CHS), 3.19 (m, 1H, CHS), 3.28 (m, 1H, H-3Gal), 3.36–3.46 (m, 2H, 2Gal, 5Gal), 3.47–3.58 (m, 2H, 6aGal, 6bGal), 3.69 (m, 1H, H-4Gal), 4.30 (d, 1H, J = 9.4 Hz Hz, H-1Gal), 4.45 (d, 1H, J = 4.5 Hz, OH), 4.58–4.73 (m, 3H, CH2N3, 6-OH), 4.82 (d, 1H, J = 5.4 Hz, OH), 5.06 (d, 1H, J = 5.7 Hz, OH), 5.35 (s, 2H, CH2O), 7.42 (dd, 1H, J = 4.2 Hz, J = 4.9 Hz, H-7Quin), 7.50–7.57 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 8.32 (dd, 1H, J = 1.8 Hz, J = 8.3 Hz, H-4Quin), 8.34 (s, 1H, H-5Triaz), 8.83 (dd, 1H, J = 1.8 Hz, J = 4.1 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 29.66 (CH2S), 50.04 (CH2N3), 60.82, 61.91 (C-6Gal, CH2O), 68.57, 69.65, 74.64, 79.36 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 85.80 (C-1Gal), 110.07 (C-7Quin), 120.06 (C-5Quin), 121.92 (C-3Quin), 125.37 (C-5Triaz), 126.83 (C-6Quin), 129.11 (C-4aQuin), 135.89 (C-4Quin), 139.71 (C-8aQuin), 142.31 (C-4Triaz), 149.03 (C-2Quin), 153.88 (C-8Quin); HRMS (ESI-TOF): calcd for C20H25N4O6S ([M + H]+): m/z 449.1495; found: m/z 449.1494.
Glycoconjugate57: Starting from sugar derivative21and 8-HQ derivative40, the product was obtained as a brown solid (87% yield), m.p.: 91–95 °C; [α]23D = 5.0 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 2.63 (s, 3H, CH3), 2.98–3.23 (m, 6H, H-2Glu, H-3Glu, H-4Glu H-5Glu, CH2S), 3.44 (m, 1H, 6aGlu), 3.72 (m, 1H, 6bGlu), 4.38 (d, 1H, J = 9.6 Hz, H-1Glu), 4.55–4.73 (m, 3H, CH2N3, 6-OH), 4.95 (d, 1H, J = 5.3 Hz, OH), 5.03 (d, 1H, J = 4.8 Hz, OH), 5.20 (d, 1H, J = 5.8 Hz, OH), 5.35 (s, 2H, CH2O), 7.37 (dd, 1H, J = 1.7 Hz, J = 8.1 Hz, H-7Quin), 7.39–7.44 (m, 2H, H-3Quin, H-6Quin), 7.47 (dd, 1H, J = 1.7 Hz, J = 7.4 Hz, H-5Quin), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin), 8.33 (s, 1H, H-5Triaz); 13C NMR (100 MHz, DMSO): δ 24.83 (CH3), 29.70 (CH2S), 49.82 (CH2N), 61.19, 61.68 (C-6Glu, CH2O), 69.96, 72.85, 78.01, 80.91 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 85.30 (C-1Glu), 110.21 (C-7Quin), 119.74 (C-5Quin), 122.36 (C-3Quin), 125.34 (C-5Triaz), 125.57 (C-6Quin), 127.24 (C-4aQuin), 135.88 (C-4Quin), 139.10 (C-8aQuin), 142.21 (C-4Triaz), 153.28 (C-2Quin), 157.16 (C-8Quin); HRMS (ESI-TOF): calcd for C21H27N4O6S ([M + H]+): m/z 463.1651; found: m/z 463.1651.
Glycoconjugate58: Starting from sugar derivative22and 8-HQ derivative40, the product was obtained as a brown solid (88% yield), m.p.: 83–87 °C; [α]23D = 10.2 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 2.63 (s, 3H, CH3), 3.05 (m, 1H, CHS), 3.18 (m, 1H, CHS), 3.28 (m, 1H, H-3Gal), 3.36–3.46 (m, 2H, H-2Gal, H-5Gal), 3.48–3.58 (m, 2H, 6aGal, 6bGal), 3.68 (m, 1H, H-4Gal), 4.30 (d, 1H, J = 9.5 Hz, H-1Gal), 4.47 (d, 1H, J = 4.6 Hz, OH), 4.57–4.72 (m, 3H, CH2N3, 6-OH), 4.86 (d, 1H, J = 5.6 Hz, OH), 5.09 (d, 1H, J = 5.8 Hz, OH), 5.34 (s, 2H, CH2O), 7.37 (dd, 1H, J = 1.8 Hz, J = 7.4 Hz, H-7Quin), 7.40–7.46 (m, 2H, H-3Quin, H-6Quin), 7.47 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, H-5Quin), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin), 8.33 (s, 1H, H-5Triaz); 13C NMR (100 MHz, DMSO): δ 24.97 (CH3), 29.67 (CH2S), 50.01 (CH2N3), 60.77, 61.71 (C-6Gal, CH2O), 68.52, 69.60, 74.63, 79.36 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 85.84 (C-1Gal), 110.23 (C-7Quin), 119.85 (C-5Quin), 122.52 (C-3Quin), 125.43, 125.72 (C-5Triaz, C-6Quin), 127.36 (C-4aQuin), 136.03 (C-4Quin), 139.19 (C-8aQuin), 142.34 (C-4Triaz), 153.39 (C-2Quin), 157.32 (C-8Quin); HRMS (ESI-TOF): calcd for C21H27N4O6S ([M + H]+): m/z 463.1651; found: m/z 463.1654.
Glycoconjugate59: Starting from sugar derivative23and 8-HQ derivative41, the product was obtained as a brown oil (94% yield), [α]23D = 36.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 2.00, 2.02, 2.06 (4s, 12H, CH3CO), 2.63 (p, 2H, J = 6.3 Hz, CH2), 3.67 (ddd, 1H, J = 2.2 Hz, J = 4.6 Hz, J = 10.0 Hz, H-5Glu), 3.85 and 4.05 (qAB, 2H, J = 14.3 Hz, CH2S), 4.07 (dd, 1H, J = 2.2 Hz, J = 12.4 Hz, H-6aGlu), 4.20 (dd, 1H, J = 4.6 Hz, J = 12.4 Hz, H-6bGlu), 4.24 (t, 2H, J = 5.9 Hz, CH2O), 4.60 (d, 1H, J = 10.1 Hz, H-1Glu), 4.73 (t, 2H, J = 6.7 Hz, CH2N), 5.02 (dd~t, 1H, J = 9.3 Hz, J = 10.1 Hz, H-2Glu), 5.08 (dd~t, 1H, J = 9.4 Hz, J = 10.0 Hz, H-4Glu), 5.19 (dd~t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3Glu), 7.05 (dd, 1H, J = 2.1 Hz, J = 6.7 Hz, H-7Quin), 7.39–7.52 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.67 (s, 1H, H-5Triaz), 8.17 (dd, 1H, J = 1.6 Hz, J = 8.3 Hz, H-4Quin), 8.96 (dd, 1H, J = 1.6 Hz, J = 4.0 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.62, 20.66, 20.77 (CH3CO), 24.39 (CH2), 29.61 (CH2S) 47.28 (CH2N), 61.85 (C-6Glu), 65.19 (CH2O), 68.15, 69.89, 73.82, 75.76 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 82.75 (C-1Glu), 109.40 (C-7Quin), 120.34 (C-5Quin), 121.74 (C-3Quin), 123.03 (C-5Triaz), 126.71 (C-6Quin), 129.56 (C-4aQuin), 136.08 (C-4Quin), 140.30 (C-8aQuin), 144.53 (C-4Triaz), 149.41 (C-2Quin), 154.20 (C-8Quin), 169.40, 169.81, 170.14, 170.65 (CH3CO); HRMS (ESI-TOF): calcd for C29H35N4O10S ([M + H]+): m/z 631.2074; found: m/z 631.2070.
Glycoconjugate60: Starting from sugar derivative24and 8-HQ derivative41, the product was obtained as a beige solid (79% yield), m.p.: 56–58 °C; [α]25D = −27.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.97, 1.99, 2.02, 2.14 (4s, 12H, CH3CO), 2.63 (p, 2H, J = 6.3 Hz, CH2), 3.80 (m, 1H, H-5Gal), 3.88 and 4.07 (qAB, 2H, J = 14.2 Hz, CH2S), 3.96–4.07 (m, 2H, CH2O), 4.16–4.31 (m, 2H, H-6aGal, H-6bGal), 4.55 (d, 1H, J = 10.0 Hz, H-1Gal), 4.73 (t, 2H, J = 6.7 Hz, CH2N), 5.00 (dd~t, 1H, J = 3.4 Hz, J = 10.0 Hz, H-3Gal), 5.22 (dd~t, 1H, J = 10.0 Hz, J = 10.0 Hz, H-2Gal), 5.38 (d, 1H, J = 2.9 Hz, H-4Gal), 7.06 (dd, 1H, J = 1.6 Hz, J = 7.0 Hz, H-7Quin), 7.40–7.51 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.66 (s, 1H, H-5Triaz), 8.16 (dd, 1H, J = 1.5 Hz, J = 8.3 Hz, H-4Quin), 8.96 (dd, 1H, J = 1.4 Hz, J = 4.0 Hz, H-2Quin); 13C NMR (100 MHz, CDCl3): δ 20.58, 20.67, 20.69, 20.74 (CH3CO), 24.24 (CH2), 29.65 (CH2S) 47.25 (CH2N), 61.42 (C-6Gal), 65.19 (CH2O), 67.30, 71.78, 74.45, 77.23 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 83.04 (C-1Gal), 109.44 (C-7Quin), 120.38 (C-5Quin), 121.77 (C-3Quin), 123.04 (C-5Triaz), 126.74 (C-6Quin), 129.59 (C-4aQuin), 136.06 (C-4Quin), 140.35 (C-8aQuin), 144.38 (C-4Triaz), 149.43 (C-2Quin), 154.26 (C-8Quin), 169.58, 169.97, 170.21, 170.31 (CH3CO); HRMS (ESI-TOF): calcd for C29H35N4O10S ([M + H]+): m/z 631.2074; found: m/z 631.2076.
Glycoconjugate61: Starting from sugar derivative23and 8-HQ derivative42, the product was obtained as a brown solid (77% yield), m.p.: 48–49 °C; [α]23D = 36.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.97, 2.00, 2.02, 2.06 (4s, 12H, CH3CO), 2.56 (p, 2H, J = 6.3 Hz, CH2), 2.79 (s, 3H, CH3), 3.65 (ddd, 1H, J = 2.1 Hz, J = 4.5 Hz, J = 10.0 Hz, H-5Glu), 3.85 and 4.05 (qAB, 2H, J = 14.3 Hz, CH2S), 4.06 (dd, 1H, J = 2.1 Hz, J = 12.4 Hz, H-6aGlu), 4.19 (dd, 1H, J = 4.5 Hz, J = 12.4 Hz, H-6bGlu), 4.24 (t, 2H, J = 5.4 Hz, CH2O), 4.60 (d, 1H, J = 10.1 Hz, H-1Glu), 4.74 (t, 2H, J = 6.7 Hz, CH2N), 5.01 (dd~t, 1H, J = 9.3 Hz, J = 10.1 Hz, H-2Glu), 5.08 (dd~t, 1H, J = 9.4 Hz, J = 10.0 Hz, H-4Glu), 5.18 (dd~t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3Glu), 7.04 (dd, 1H, J = 2.8 Hz, J = 6.1 Hz, H-7Quin), 7.30–7.43 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.74 (s, 1H, H-5Triaz), 8.04 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.61, 20.65, 20.77 (CH3CO), 24.42 (CH2), 25.75 (CH3), 29.63 (CH2S) 47.20 (CH2N), 61.84 (C-6Glu), 65.34 (CH2O), 68.14, 69.91, 73.83, 75.72 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 82.74 (C-1Glu), 110.06 (C-7Quin), 120.32 (C-5Quin), 122.63 (C-3Quin), 123.08 (C-5Triaz), 125.70 (C-6Quin), 127.76 (C-4aQuin), 136.19 (C-4Quin), 139.91 (C-8aQuin), 144.47 (C-4Triaz), 153.64 (C-2Quin), 158.23 (C-8Quin), 169.40, 169.77, 170.14, 170.65 (CH3CO); HRMS (ESI-TOF): calcd for C30H37N4O10S ([M + H]+): m/z 645.2230; found: m/z 645.2233.
Glycoconjugate62: Starting from sugar derivative24and 8-HQ derivative42, the product was obtained as a beige solid (80% yield), m.p.: 56–58 °C; [α]24D = −26.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.97, 1.98, 2.02, 2.14 (4s, 12H, CH3CO), 2.60 (p, 2H, J = 6.3 Hz, CH2), 2.79 (s, 3H, CH3), 3.78 (m, 1H, H-5Gal), 3.88 and 4.07 (qAB, 2H, J = 14.1 Hz, CH2S), 3.96–4.10 (m, 2H, CH2O), 4.15–4.31 (m, 2H, H-6aGal, H-6bGal), 4.54 (d, 1H, J = 10.0 Hz, H-1Gal), 4.75 (t, 2H, J = 6.7 Hz, CH2N), 5.00 (dd~t, 1H, J = 3.2 Hz, J = 10.0 Hz, H-3Gal), 5.22 (dd~t, 1H, J = 10.0 Hz, J = 10.0 Hz, H-2Gal), 5.37 (d, 1H, J = 3.2 Hz, H-4Gal), 7.04 (m, 1H, H-7Quin), 7.33 (d, 1H, J = 8.4 Hz, H-3Quin), 7.36–7.43 (m, 2H, H-5Quin, H-6Quin), 7.72 (s, 1H, H-5Triaz), 8.04 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, CDCl3): δ 20.58, 20.62, 20.68, 20.74 (CH3CO), 24.26 (CH2), 25.70 (CH3), 29.67 (CH2S) 47.18 (CH2N), 61.41 (C-6Gal), 65.32 (CH2O), 67.30, 67.32, 71.78, 74.42 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 83.02 (C-1Gal), 110.07 (C-7Quin), 120.34 (C-5Quin), 122.65 (C-3Quin), 123.07 (C-5Triaz), 125.73 (C-6Quin), 127.80 (C-4aQuin), 136.20 (C-4Quin), 139.93 (C-8aQuin), 144.31 (C-4Triaz), 153.70 (C-2Quin), 158.25 (C-8Quin), 169.58, 169.97, 170.22, 170.30 (CH3CO); HRMS (ESI-TOF): calcd for C30H37N4O10S ([M + H]+): m/z 645.2230; found: m/z 645.2233.
Glycoconjugate63: Starting from sugar derivative25and 8-HQ derivative41, the product was obtained as a beige solid (81% yield), m.p.: 59–62 °C; [α]25D = −51.0 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 2.41 (p, 2H, J = 6.5 Hz, CH2), 3.02–3.10 (m, 2H, H-3Glu, H-5Glu), 3.11–3.19 (m, 2H, H-2Glu, H-6aGlu), 3.45 (m, 1H, H-6bGlu), 3.73 (m, 1H, H-4Glu), 3.85 and 3.98 (qAB, 2H, J = 14.2, CH2S), 4.19 (t, 2H, J = 6.1 Hz CH2O), 4.27 (d, 1H, J = 9.6 Hz, H-1Glu), 4.61 (t, 2H, J = 7.0 Hz, CH2N), 4.75 (t, 1H, J = 5.7 Hz, 6-OH), 4.97 (d, 1H, J = 5.3 Hz, OH), 5.00 (d, 1H, J = 4.9 Hz, OH), 5.17 (d, 1H, J = 5.9 Hz, OH), 7.20 (dd, 1H, J = 1.8 Hz, J = 7.1 Hz, H-7Quin), 7.48–7.54 (m, 2H, H-5Quin, H-6Quin), 7.59 (dd, 1H, J = 4.1 Hz, J = 8.3 Hz, H-3Quin), 8.20 (s, 1H, H-5Triaz), 8.33 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4Quin), 8.90 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 23.16 (CH2), 29.61 (CH2S), 46.59 (CH2N), 61.35 (C-6Glu), 65.38 (CH2O), 70.14, 73.08, 78.16, 81.06 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 84.26 (C-1Glu), 109.80 (C-7Quin), 119.89 (C-5Quin), 121.86 (C-3Quin), 123.52 (C-5Triaz), 126.82 (C-6Quin), 129.05 (C-4aQuin), 135.85 (C-4Quin), 139.72 (C-8aQuin), 144.39 (C-4Triaz), 149.07 (C-2Quin), 154.15 (C-8Quin); HRMS (ESI-TOF): calcd for C21H27N4O6S ([M + H]+): m/z 463.1651; found: m/z 463.1653.
Glycoconjugate64: Starting from sugar derivative26and 8-HQ derivative41, the product was obtained as a white solid (89% yield), m.p.: 150–153 °C; [α]26D = −30.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 2.42 (p, 2H, J = 6.5 Hz, CH2), 3.25 (m, 1H, H-3Gal), 3.34–3.46 (m, 2H, H-2Gal, H-5Gal), 3.45–3.58 (m, 2H, H-6aGal, H-6bGal), 3.65 (m, 1H, H-4Gal), 3.84 and 3.96 (qAB, 2H, J = 14.1 Hz, CH2S), 4.13–4.24 (m, 2H, CH2O), 4.20 (d, 1H, J = 9.5 Hz, H-1Gal), 4.42 (d, 1H, J = 3.8 Hz, OH), 4.61 (t, 2H, J = 7.0 Hz, CH2N), 4.72 (bs, 1H, 6-OH), 4.78 (d, 1H, J = 4.4 Hz, OH), 5.00 (d, 1H, J = 5.5 Hz, OH), 7.19 (dd, 1H, J = 2.2 Hz, J = 6.8 Hz, H-7Quin), 7.47–7.54 (m, 2H, H-5Quin, H-6Quin), 7.59 (dd, 1H, J = 4.2 Hz, J = 8.3 Hz, H-3Quin), 8.18 (s, 1H, H-5Triaz), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4Quin), 8.90 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2Quin); 13C NMR (100 MHz, DMSO): δ 23.08 (CH2), 29.59 (CH2S), 46.58 (CH2N), 60.86 (C-6Gal), 65.34 (CH2O), 68.60, 69.84, 74.64, 79.38 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.70 (C-1Gal), 109.78 (C-7Quin), 119.90 (C-5Quin), 121.87 (C-3Quin), 123.53 (C-5Triaz), 126.82 (C-6Quin), 129.05 (C-4aQuin), 135.85 (C-4Quin), 139.73 (C-8aQuin), 144.41 (C-4Triaz), 149.09 (C-2Quin), 154.16 (C-8Quin); HRMS (ESI-TOF): calcd for C21H27N4O6S ([M + H]+): m/z 463.1651; found: m/z 463.1651.
Glycoconjugate65: Starting from sugar derivative25and 8-HQ derivative42, the product was obtained as a brown solid (91% yield), m.p.: 55–58 °C; [α]26D = −40.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 2.40 (p, 2H, J = 6.6 Hz, CH2), 2.67 (s, 3H, CH3), 2.99–3.19 (m, 4H, H-2Glu, H-3Glu, H-4Glu, H-5Glu), 3.43 (m, 1H, H-6aGlu), 3.71 (m, 1H, H-6bGlu), 3.84 and 3.98 (qAB, 2H, J = 14.1 Hz, CH2S), 4.19 (t, 2H, J = 6.2 Hz, CH2O), 4.26 (d, 1H, J = 9.6 Hz, H-1Glu), 4.61 (t, 2H, J = 7.0 Hz, CH2N), 4.69 (t, 1H, J = 5.8 Hz, 6-OH), 4.94 (d, 1H, J = 5.1 Hz, OH), 5.01 (d, 1H, J = 4.7 Hz, OH), 5.15 (d, 1H, J = 5.8 Hz, OH), 7.15 (dd, 1H, J = 1.3 Hz, J = 7.5 Hz, H-7Quin), 7.38–7.49 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 8.17 (s, 1H, H-5Triaz), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, DMSO): δ 23.10 (CH2), 25.07 (CH3), 29.54 (CH2S), 46.58 (CH2N), 61.31 (C-6Glu), 65.54 (CH2O), 70.13, 73.09, 78.16, 81.06 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 84.21 (C-1Glu), 110.33 (C-7Quin), 119.83 (C-5Quin), 122.47 (C-3Quin), 123.45 (C-5Triaz), 125.75 (C-6Quin), 127.33 (C-4aQuin), 136.00 (C-4Quin), 139.31 (C-8aQuin), 144.36 (C-4Triaz), 153.67 (C-2Quin), 157.32 (C-8Quin); HRMS (ESI-TOF): calcd for C22H29N4O6S ([M + H]+): m/z 477.1808; found: m/z 477.1806.
Glycoconjugate66: Starting from sugar derivative26and 8-HQ derivative42, the product was obtained as a beige solid (90% yield), m.p.: 83–87 °C; [α]23D = 10.2 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 2.41 (p, 2H, J = 6.3 Hz, CH2), 3.43 (s, 3H, CH3), 3.23 (m, 1H, H-3Gal), 3.34–3.42 (m, 2H, H-2Gal, H-5Gal), 3.43–3.58 (m, 2H, H-6aGal, H-6bGal), 3.65 (m, 1H, H-4Gal), 3.83 and 3.96 (qAB, 2H, J = 14.1 Hz, CH2S), 4.12–4.24 (m, 2H, CH2O), 4.19 (d, 1H, J = 9.4 Hz, H-1Gal), 4.41 (d, 1H, J = 4.0 Hz, OH), 4.61 (t, 2H, J = 7.0 Hz, CH2N), 4.68 (bs, 1H, 6-OH), 4.77 (d, 1H, J = 4.7 Hz, OH), 4.99 (d, 1H, J = 5.6 Hz, OH), 7.16 (dd, 1H, J = 1.3 Hz, J = 7.6 Hz, H-7Quin), 7.38–7.45 (m, 2H, H-3Quin, H-6Quin), 7.47 (dd, 1H, J = 1.4 Hz, J = 8.2 Hz, H-5Quin), 8.16 (s, 1H, H-5Triaz), 8.19 (d, 1H, J = 8.4 Hz, H-4Quin); 13C NMR (100 MHz, DMSO): δ 23.02 (CH2), 25.08 (CH3), 29.52 (CH2S), 46.56 (CH2N), 60.81 (C-6Gal), 65.50 (CH2O), 68.57, 69.83, 74.64, 79.36 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 84.65 (C-1Gal), 110.31 (C-7Quin), 119.83 (C-5Quin), 122.47 (C-3Quin), 123.43 (C-5Triaz), 125.73 (C-6Quin), 127.33 (C-4aQuin), 135.99 (C-4Quin), 139.31 (C-8aQuin), 144.37 (C-4Triaz), 153.67 (C-2Quin), 157.33 (C-8Quin); HRMS (ESI-TOF): calcd for C22H29N4O6S ([M + H]+): m/z 477.1808; found: m/z 477.1807.
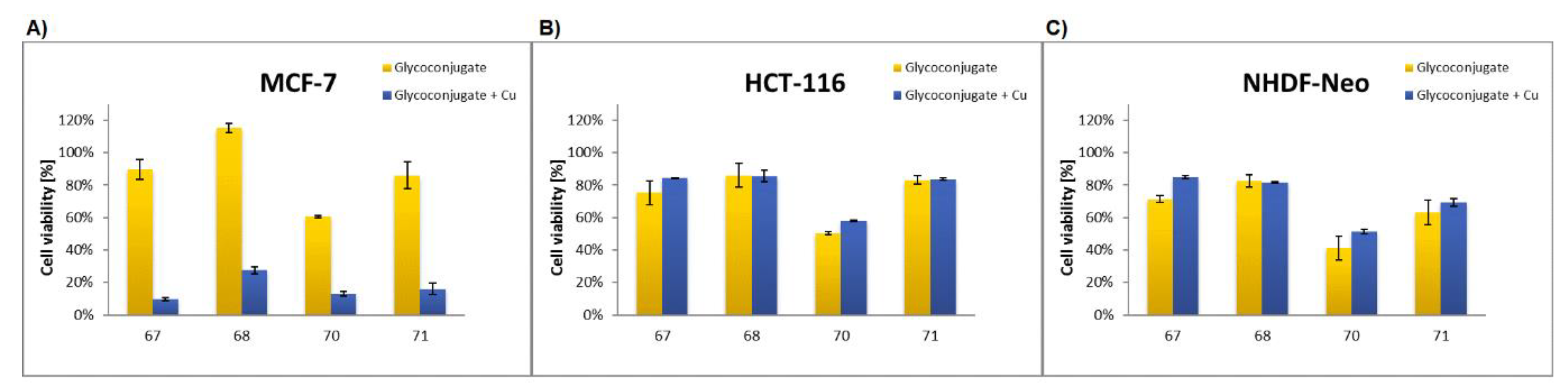
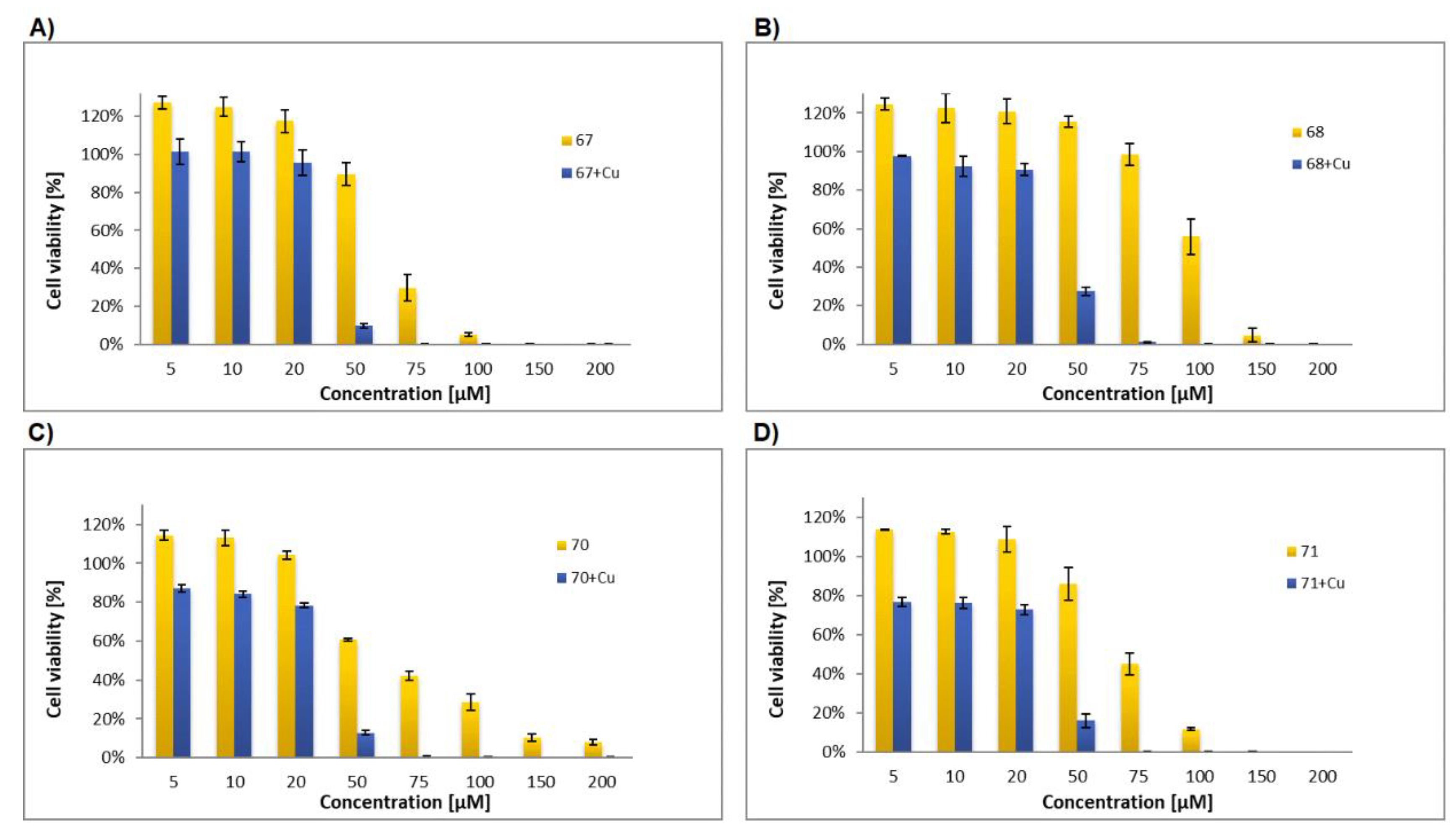
Glycoconjugate67: Starting from sugar derivative33and 8-HQ derivative39, the product was obtained as a brown solid (84% yield), m.p.: 114–115 °C; [α]20D = −2.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.97, 2.00, 2.00, 2.02 (4s, 12H, CH3CO), 3.80 (ddd, 1H, J = 2.2 Hz, J = 4.4 Hz, J = 10.1 Hz, H-5Glu), 4.07 (dd, 1H, J = 2.2 Hz, J = 12.4 Hz, H-6aGlu), 4.22 (dd, 1H, J = 4.4 Hz, J = 12.4 Hz, H-6bGlu), 5.05–5.20 (m, 4H, H-2Glu, H-4Glu, CH2O), 5.29 (dd~t, 1H, J = 9.3 Hz, J = 9.3 Hz, H-3Glu), 5.40 (s, 2H, CH2O), 5.59 (d, 1H, J = 10.4 Hz, H-1Glu), 6.89 (d, 1H, J = 8.4 Hz, H-3Pyr), 7.34 (d, 1H, J = 6.6 Hz, H-7Quin), 7.44–7.59 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.76 (d, 1H, J = 8.4 Hz, H-4Pyr), 7.97 (s, 1H, H-5Triaz), 8.25 (d, 1H, J = 7.7 Hz, H-4Quin), 8.41 (s, 1H, H-6Pyr), 8.84 (bs, 1H, H-2Quin), 10.84 (bs, 1H, NH); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.62, 20.71, 20.71 (CH3CO), 52.80 (CH2N), 61. 87, 62.04 (CH2O, C-6Glu), 68.20, 69.47, 74.11, 75.83 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 82.22 (C-1Glu), 109.56 (C-7Quin), 120.45 (C-5Quin), 122.10 (C-3Quin), 123.27 (C-5Triaz), 125.92 (CPyr), 127.29 (CPyr), 128.09 (C-6Quin), 129.73 (C-4aQuin), 132.80 (C-7Quin), 136.99 (C-4Quin), 139.54 (C-7Quin), 141.30 (C-8aQuin), 142.92 (C-4Triaz), 148.63 (C-7Quin), 149.61 (C-2Quin), 153.69 (C-8Quin), 163.96 (C=O), 169.44, 169.53, 170.11, 170.65 (CH3CO); HRMS (ESI-TOF): calcd for C33H35N6O11S ([M + H]+): m/z 732.2085; found: m/z 732.2084.
Glycoconjugate68: Starting from sugar derivative34and 8-HQ derivative39, the product was obtained as a beige solid (83% yield), m.p.: 128–129 °C; [α]21D = 9.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.94, 1.98, 2,01, 2.15 (4s, 12H, CH3CO), 4.01 (m, 1H, H-5Gal), 4.05–4.10 (m, 2H, H-6aGal, H-6bGal), 5.11 (s, 2H, CH2O), 5.14 (dd, 1H, J = 3.4 Hz, J = 10.1 Hz, H-3Gal), 5.36 (dd~t, 1H, J = 10.1 Hz, J = 10.2 Hz, H-2Gal), 5.40 (s, 2H, CH2N), 5.45 (dd, 1H, J = 0.7 Hz, J = 3.4 Hz, H-4Gal), 5.58 (d, 1H, J = 10.2 Hz, H-1Gal), 7.00 (d, 1H, J = 8.7 Hz, H-3Pyr), 7.33 (d, 1H, J = 7.4 Hz, H-7Quin), 7.44–7.58 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.81 (dd, 1H, J = 2.3 Hz, J = 8.7 Hz, H-4Pyr), 7.97 (s, 1H, H-5Triaz), 8.24 (d, 1H, J = 8.3 Hz, H-4Quin), 8.43 (d, 1H, J = 1.9 Hz, H-6Pyr), 8.32 (d, 1H, J = 3.0 Hz, H-2Quin), 10.77 (bs, 1H, NH); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.64, 20.68, 20.80 (CH3CO), 52.83 (CH2N), 61. 23, 62.07 (CH2O, C-6Gal), 66.84, 67.29, 72.04, 74.46 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 82.75 (C-1Gal), 109.57 (C-7Quin), 120.49 (C-5Quin), 122.08 (C-3Quin), 123.29 (C-5Triaz), 125.85 (CPyr), 127.24 (CPyr), 128.21 (C-6Quin), 129.73 (C-4aQuin), 132.76 (C-7Quin), 136.96 (C-4Quin), 139.55 (C-7Quin), 141.36 (C-8aQuin), 143.02 (C-4Triaz), 148.65 (C-7Quin), 149.93 (C-2Quin), 153.67 (C-8Quin), 163.91 (C=O), 169.72, 169.98, 170.24, 170.33 (CH3CO); HRMS (ESI-TOF): calcd for C33H35N6O11S ([M + H]+): m/z 723.2085; found: m/z 723.2086.
Glycoconjugate69: Starting from sugar derivative35and 8-HQ derivative39, the product was obtained as a brown solid (77% yield), m.p.: 143–144 °C; [α]22D = −24.0 (c = 0.5, H2O); 1H NMR (400 MHz, DMSO): δ 3.13 (m, 1H, H-2Glu), 3.21–3.29 (m, 2H, H-3Glu, H-4Glu), 3.37–3.50 (m, 2H, H-6aGlu, H-6bGlu), 3.66 (m, 1H, H-5Glu), 4.51 (t, 1H, J = 5.7 Hz, 6-OH), 5.00 (d, 1H, J = 5.3 Hz, OH), 5.07 (d, 1H, J = 9.9 Hz, H-1Glu),5.13 (d, 1H, J = 4.9 Hz, OH), 5.36 (d, 1H, J = 6.1 Hz, OH), 5.39 (t, 2H, J = 6.0 Hz, CH2O), 5.42 (t, 2H, J = 6.9 Hz, CH2N), 7.40 (d, 1H, J = 8.6 Hz, H-3Pyr), 7.44 (dd, 1H, J = 4.4 Hz, J = 8.9 Hz, H-7Quin), 7.50–7.58 (m, 3H, H-3Quin, H-5Quin, H-6Quin), 7.92 (dd, 1H, J = 2.6 Hz, J = 8.6 Hz, H-4Pyr), 8.32 (dd, 1H, J = 1.8 Hz, J = 8.3 Hz, H-4Quin), 8.35 (s, 1H, H-5Triaz), 8.63 (d, 1H, J = 2.6 Hz, H-6Pyr), 8.84 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2Quin), 10.73 (s, 1H, NH); 13C NMR (100 MHz, DMSO): δ 52.10 (CH2N), 60. 83, 61.77 (CH2O, C-6Glu), 69.71, 72.39, 78.28, 81.05 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 84.60 (C-1Glu), 109.94 (C-7Quin), 119.98 (C-5Quin), 121.85 (C-3Quin), 122.17 (C-5Triaz), 126.51 (CPyr), 126.74 (CPyr), 127.76 (C-6Quin), 129.06 (C-4aQuin), 132.35 (C-7Quin), 135.77 (C-4Quin), 139.73 (C-7Quin), 140.29 (C-8aQuin), 142.49 (C-4Triaz), 148.98 (C-7Quin), 152.52 (C-2Quin), 153.87 (C-8Quin), 164.69 (C=O); HRMS (ESI-TOF): calcd for C25H27N6O7S ([M + H]+): m/z 555.1662; found: m/z 555.1667.
Glycoconjugate70: Starting from sugar derivative36and 8-HQ derivative41, the product was obtained as a white solid (72% yield), m.p.: 93–95 °C; [α]21D = −3.0 (c = 1, CHCl3); 1H NMR (400 MHz, DMSO): δ 1.96, 1.97, 1.98, 2.00 (4s, 12H, CH3CO), 2.43 (bs, 2H, CH2), 4.02 (m, 1H, H-5Glu), 4.10–4.17 (m, 2H, H-6aGlu, H-6bGlu), 4.21 (bs, 2H, CH2O), 4.67 (t, 2H, J = 6.0 Hz, CH2N), 4.93–5.02 (m, 2H, H-2Glu, H-4Glu), 5.22 (s, 2H, CH2OCO), 5.41 (dd~t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3Glu), 5.69 (d, 1H, J = 10.3 Hz, H-1Glu), 7.21 (d, 1H, J = 8.0 Hz, H-7Quin), 7.37 (d, 1H, J = 8.7 Hz, H-3Pyr), 7.50 (dd~t, 1H, J = 7.5 Hz, J = 8.0 Hz, H-3Quin), 7.47–7.64 (m, 2H, H-5Quin, H-6Quin), 7.83 (dd, 1H, J = 2.4 Hz, J = 8.7 Hz, H-4Pyr), 8.32–8.40 (m, 2H, H-4Quin, H-6Pyr), 8.54 (s, 1H, H-5Triaz), 8.91 (bs, 1H, H-2Quin), 10.00 (s, 1H, NH); 13C NMR (100 MHz, DMSO): δ 20.19, 20.24, 20.28, 20.24 (CH3CO), 29.51 (CH2), 46.63 (CH2N), 57.63 (CH2OCO), 61.73 (C-6Glu), 65.36 (CH2O), 67.94, 69.33, 72.91, 74.41 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 81.26 (C-1Glu), 109.82 (C-7Quin), 119.92 (C-5Quin), 121.81 (C-3Quin), 123.05 (C-5Triaz), 125.00 (CPyr), 126.60 (CPyr), 126.78 (C-6Quin), 128.98 (C-4aQuin), 133.84 (CPyr), 135.92 (C-4Quin), 139.43 (CPyr), 139.68 (C-8aQuin), 141.91 (C-4Triaz), 148.01 (CPyr), 148.84 (C-2Quin), 153.10 (C-8Quin), 153.99 (C=O), 168.99, 169.18, 169.38, 169.80 (CH3CO); HRMS (ESI-TOF): calcd for C35H39N6O12S ([M + H]+): m/z 767.2347; found: m/z 767.2344.
Glycoconjugate71: Starting from sugar derivative37and 8-HQ derivative41, the product was obtained as a white solid (73% yield), m.p.: 97–99 °C; [α]21D = 9.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO): δ 1.94, 1.95, 2.00, 2.14 (4s, 12H, CH3CO), 2.43 (bs, 2H, CH2), 3.97–4.07 (m, 2H, H-6aGal, H-6bGal), 4.21 (bs, 2H, CH2O), 4.35 (m, 1H, H-5Gal), 4.67 (t, 2H, J = 6.5 Hz, CH2N), 5.13 (dd~t, 1H, J = 9.9 Hz, J = 10.2 Hz, H-2Gal), 5.22 (s, 2H, CH2OCO), 5.30–5.38 (m, 2H, H-3Gal, H-4Gal), 5.65 (d, 1H, J = 10.2 Hz, H-1Gal), 7.21 (d, 1H, J = 8.4 Hz, H-7Quin), 7.38 (d, 1H, J = 8.7 Hz, H-3Pyr), 7.50 (dd~t, 1H, J = 7.5 Hz, J = 8.1 Hz, H-3Quin), 7.50–7.65 (m, 2H, H-5Quin, H-6Quin), 7.84 (dd, 1H, J = 2.4 Hz, J = 8.7 Hz, H-4Pyr), 8.32–8.40 (m, 2H, H-4Quin, H-6Pyr), 8.54 (s, 1H, H-5Triaz), 8.90 (bs, 1H, H-2Quin), 10.00 (s, 1H, NH); 13C NMR (100 MHz, DMSO): δ 20.33, 20.39, 20.41, 20.43 (CH3CO), 29.60 (CH2), 46.72 (CH2N), 57.72 (CH2OCO), 61.51 (C-6Gal), 65.44 (CH2O), 66.80, 67.55, 71.01, 73.66 (C-2Gal, C-3Gal, C-4Gal, C-5Gal), 81.77 (C-1Gal), 109.89 (C-7Quin), 120.00 (C-5Quin), 121.91 (C-3Quin), 123.04 (C-5Triaz), 125.08 (CPyr), 126.67 (CPyr), 126.85 (C-6Quin), 129.08 (C-4aQuin), 133.87 (CPyr), 135.95 (C-4Quin), 139.36 (CPyr), 139.78 (C-8aQuin), 142.00 (C-4Triaz), 148.29 (CPyr), 148.90 (C-2Quin), 153.19 (C-8Quin), 154.18 (C=O), 169.31, 169.39, 169.75, 169.94 (CH3CO); HRMS (ESI-TOF): calcd for C35H39N6O12S ([M + H]+): m/z 767.2347; found: m/z 767.2345.
Glycoconjugate72: Starting from sugar derivative38and 8-HQ derivative41, the product was obtained as a white solid (78% yield), m.p.: 152–154 °C; [α]22D = −36.0 (c = 1.0, CH3OH); 1H NMR (400 MHz, DMSO): δ 2.43 (p, 2H, J = 6.3 Hz, CH2),3.09–3.19 (m, 2H, H-2Glu, H-4Glu), 3.21–3.29 (m, 2H, H-3Glu, H-5Glu), 3.44 (m, 1H, H-6aGlu), 3.66 (m, 1H, H-6bGlu), 4.19 (t, 2H, J = 6.0 Hz, CH2O), 4.51 (t, 1H, J = 5.7 Hz, 6-OH), 4.67 (t, 2H, J = 6.9 Hz, CH2N), 4.99 (d, 1H, J = 5.3 Hz, OH), 5.03 (d, 1H, J = 9.9 Hz, H-1Glu), 5.11 (d, 1H, J = 4.8 Hz, OH), 5.21 (s, 2H, CH2OCO), 5.34 (d, 1H, J = 6.1 Hz, OH), 7.19 (dd, 1H, J = 1.7 Hz, J = 7.1 Hz, H-7Quin), 7.36 (d, 1H, J = 8.7 Hz, H-3Pyr), 7.45–7.60 (m, 2H, H-5Quin, H-6Quin), 7.56 (dd, 1H, J = 4.0 Hz, J = 8.3 Hz, H-3Quin), 7.76 (dd, 1H, J = 2.1 Hz, J = 8.7 Hz, H-4Pyr), 8.32 (dd, 1H, J = 1.3 Hz, J = 7.0 Hz, H-4Quin), 8.37 (s, 1H, H-5Triaz), 8.49 (d, 1H, J = 2.1 Hz, H-6Pyr), 8.89 (bs, 1H, H-2Quin), 9.92 (bs, 1H, NH); 13C NMR (100 MHz, DMSO): δ 29.51 (CH2), 46.62 (CH2N), 57.56 (CH2OCO), 60.76 (C-6Glu), 65.32 (CH2O), 69.65, 72.30, 78.20, 80.97 (C-2Glu, C-3Glu, C-4Glu, C-5Glu), 84.71 (C-1Glu), 109.75 (C-7Quin), 119.86 (C-5Quin), 121.77 (C-3Quin), 122.19 (C-5Triaz), 124.99 (CPyr), 126.58 (CPyr), 126.68 (C-6Quin), 128.96 (C-4aQuin), 132.91 (C-7Quin), 135.71 (C-4Quin), 139.38 (C-7Quin), 139.68 (C-8aQuin), 141.96 (C-4Triaz), 148.95 (C-7Quin), 150.91 (C-2Quin), 153.12 (C-8Quin), 154.09 (C=O); HRMS (ESI-TOF): calcd for C27H31N6O8S ([M + H]+): m/z 599.1924; found: m/z 599.1919.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}